smyd1介导的肌球蛋白重链(MHC)赖氨酸K35的单甲基化调节斑马鱼和人类ipsc来源的心肌细胞的肌小体组装和稳态。
IF 4.7
2区 医学
Q1 CARDIAC & CARDIOVASCULAR SYSTEMS
引用次数: 0
摘要
SMYD家族包括一种独特的赖氨酸甲基转移酶(KMTases),可将组蛋白和非组蛋白甲基化。在其5个成员(SMYD1-5)中,SMYD1已被确定为心脏和骨骼肌特异性KMTase,与Myosin相互作用,与Unc45b和Hsp90a协调,调节粗丝组装。然而,SMYD1协调肌球蛋白组装的确切机制在很大程度上仍然未知。在这里,我们证明SMYD1与几种肌球蛋白重链(MyHC)亚型的n端区域物理结合,并特异性催化MyHC在赖氨酸35 (K35)的单甲基化。甲基化的MyHC被正确地整合到肌瘤中,而在smyd1缺陷的斑马鱼中,未甲基化的MyHC通过泛素-蛋白酶体系统(UPS)进行降解,导致粗丝组装缺陷。尽管使用MG132抑制UPS可以恢复smyd1缺陷斑马鱼胚胎中的肌球蛋白水平,但由于缺乏K35 MyHC单甲基化,适当的粗丝组装仍然受到损害。与斑马鱼横纹肌细胞类似,smyd1介导的MyHC甲基化对于粗丝组装和人类心肌细胞的内稳态至关重要,这表明Myosin调节的保守跨物种机制在近40 年前首次被描述。现在需要进一步的研究来探索靶向这一途径在心肌病和骨骼肌疾病中的治疗潜力。本文章由计算机程序翻译,如有差异,请以英文原文为准。
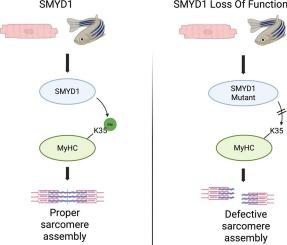
SMYD1-mediated mono-methylation of lysine K35 of sarcomeric myosin heavy chain (MHC) regulates sarcomere assembly and homeostasis in zebrafish and human iPSC-derived cardiomyocytes
The SMYD family comprises a distinct class of lysine methyltransferases (KMTases) that methylate both histone and non-histone proteins. Among its five members (SMYD1–5), SMYD1 has been identified as a cardiac and skeletal muscle-specific KMTase that interacts with Myosin, in coordination with Unc45b and Hsp90a, to regulate thick filament assembly. However, the precise mechanism by which SMYD1 orchestrates Myosin assembly remains largely unknown.
Here, we demonstrate that SMYD1 physically associates with the N-terminal region of several myosin heavy chain (MyHC) isoforms and specifically catalyzes the mono-methylation of MyHC at lysine 35 (K35). Methylated MyHC is correctly incorporated into sarcomeres, whereas unmethylated MyHC in Smyd1-deficient zebrafish undergoes degradation via the ubiquitin-proteasome system (UPS), leading to defective thick filament assembly. Although UPS inhibition with MG132 restores Myosin levels in Smyd1-deficient zebrafish embryos, proper thick filament assembly remains impaired due to the absence of K35 MyHC mono-methylation.
Similar to zebrafish striated muscle cells, SMYD1-mediated MyHC methylation is essential for thick filament assembly but also homeostasis in human cardiomyocytes, indicating a conserved cross-species mechanism of Myosin regulation, first described nearly 40 years ago. Further research is now required to explore the therapeutic potential of targeting this pathway in cardiomyopathies and skeletal muscle disorders.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
10.70
自引率
0.00%
发文量
171
审稿时长
42 days
期刊介绍:
The Journal of Molecular and Cellular Cardiology publishes work advancing knowledge of the mechanisms responsible for both normal and diseased cardiovascular function. To this end papers are published in all relevant areas. These include (but are not limited to): structural biology; genetics; proteomics; morphology; stem cells; molecular biology; metabolism; biophysics; bioengineering; computational modeling and systems analysis; electrophysiology; pharmacology and physiology. Papers are encouraged with both basic and translational approaches. The journal is directed not only to basic scientists but also to clinical cardiologists who wish to follow the rapidly advancing frontiers of basic knowledge of the heart and circulation.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


