(N)-甲醇碳-腺苷作为单胺转运体变构抑制剂:扩展的N6基团用于双视稳定
IF 5.9
2区 医学
Q1 CHEMISTRY, MEDICINAL
引用次数: 0
摘要
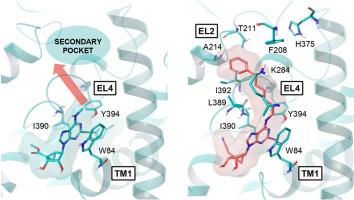
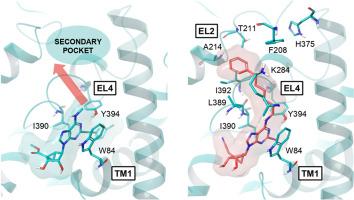
最近报道了一种人类多巴胺转运体(DAT)的低温电镜结构,该结构由一种变构抑制剂稳定,即刚性核苷mrs72921,一种tropane orthosteric抑制剂和Zn2+。我们合成了多个具有N6, C2和4 '修饰的1的North (N)-甲烷碳-腺苷类似物,以研究它们对DAT放射配体结合(增强或抑制)以及对去甲肾上腺素(NET)和血清素(SERT)转运体(通常抑制)的影响。小的N6基团提供了最明显的DAT增强(在10 μM下,N6-甲基2和N6-环丙基85 ' -乙基酯,以及N6-甲基-4 ' -氰甲基44类似物≥500%)。各种N6-(ω-苯基-烷基)基团通过访问以前未表征的DAT远端结合区来补偿1-DAT结构中显示的稳定相互作用的去除,例如N7氢键。对于双位N6-(ω-苯基-烷基)衍生物,如16和31,最佳链长为5个亚甲基,有利于5 ' -乙酯(而不是4 ' -氰乙基)和2-(芳乙基)基团。令人惊讶的是,先前注意到的2-碘基团降低的DAT活性可以由N6-(ω-苯基-烷基)基团补偿。N6-(6-苯基己基)-2-碘化合物53(5′-羟基,Ki 0.89 μM,泛抑制剂)和54(4′-氰乙基,Ki 0.40 μM)抑制DAT结合。手性支链N6基团表现出结合立体选择性。关键核苷被对接到向外的hDAT低温电镜结构上。分子动力学模拟预测了F208EL2和H375EL4(芳香保守于NET)的N6-(ω-苯基-烷基)取代基和芳香侧链的π-π相互作用,确定了扩展链的首选结合模式。因此,我们对该系列的转运体SAR进行了表征,增强或抑制正位放射配体结合和转运抑制,并进行了DAT结构预测。本文章由计算机程序翻译,如有差异,请以英文原文为准。
(N)-Methanocarba-adenosines as monoamine transporter allosteric inhibitors: Extended N6 groups for bitopic stabilization
A human dopamine transporter (DAT) cryo-EM structure was recently reported, as stabilized by an allosteric inhibitor, i.e. rigid nucleoside MRS7292 1, a tropane orthosteric inhibitor and Zn2+. We have synthesized multiple North (N)-methanocarba-adenosine analogues of 1, with N6, C2 and 4′ modifications to examine their effects on DAT radioligand binding (either enhancement or inhibition) and at the norepinephrine (NET) and serotonin (SERT) transporters (generally inhibition). Small N6 groups provided the most pronounced DAT enhancement (≥500 % at 10 μM for N6-methyl 2 and N6-cyclopropyl 8 5′-ethyl esters, and N6-methyl-4′-cyanomethyl 44 analogues). Various N6-(ω-phenyl-alkyl) groups caused binding inhibition and compensated for the removal of stabilizing interactions shown in the 1-DAT structure, e.g. an N7 H-bond, by accessing a previously uncharacterized DAT distal binding region. The optimal chain length was five methylenes for bitopic N6-(ω-phenyl-alkyl) derivatives, e.g. 16 and 31, having favored 5′-ethyl ester (but not 4′-cyanomethyl) and 2-(arylethynyl) groups. Surprisingly, the previously noted reduced DAT activity with a 2-iodo group, could be compensated by N6-(ω-phenyl-alkyl) groups. N6-(6-Phenylhexyl)-2-iodo compounds, 53 (5′-hydroxy, Ki 0.89 μM, pan-inhibitor) and 54 (4′-cyanomethyl, Ki 0.40 μM), inhibited DAT binding. Chiral, branched N6 groups displayed binding stereoselectivity. Key nucleosides were docked to outward-facing hDAT cryo-EM structures. Molecular dynamics simulation predicted π-π interactions of the N6-(ω-phenyl-alkyl) substituent and aromatic side chains of F208EL2 and H375EL4 (aromatic conserved at NET) to define a preferred binding mode for the extended chain. Thus, we characterized the transporter SAR of this series, either enhancement or inhibition of orthosteric radioligand binding, and transport inhibition, and with DAT structural predictions.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
11.70
自引率
9.00%
发文量
863
审稿时长
29 days
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


