Eduardo Pedraza, , , Andres R. Tejedor, , , Alejandro Feito, , , Francisco Gámez, , , Rosana Collepardo-Guevara, , , Eduardo Sanz*, , and , Jorge R. Espinosa*,
{"title":"用热力学积分法预测相分离蛋白的饱和浓度。","authors":"Eduardo Pedraza, , , Andres R. Tejedor, , , Alejandro Feito, , , Francisco Gámez, , , Rosana Collepardo-Guevara, , , Eduardo Sanz*, , and , Jorge R. Espinosa*, ","doi":"10.1021/acs.jctc.5c00765","DOIUrl":null,"url":null,"abstract":"<p >Phase separation of proteins and nucleic acids into biomolecular condensates contributes to the regulation of cellular compartmentalization in membrane-less environments. A key parameter controlling the onset of biomolecular condensate formation is the saturation concentration (<i>C</i><sub>sat</sub>)─the threshold concentration above which condensation takes place. While measuring <i>C</i><sub>sat</sub> for protein solutions in vitro is experimentally accessible, determining this quantity in simulations remains challenging due to the extremely low equilibrium concentrations at which many proteins phase separate. This occurs because the gold standard in simulations consists of combining a residue-resolution coarse-grained model with the Direct Coexistence simulation method, which yields poor estimates of the equilibrium concentrations of the dilute phase due to lack of statistics. In this work, we present two independent thermodynamic integration (TI) schemes which, when combined with Direct Coexistence simulations, enable accurate calculation of saturation concentrations and phase diagrams─facilitating direct comparison with experimental measurements across a wide range of conditions. Our methods, combined with the Mpipi-Recharged residue-resolution model, accurately estimate <i>C</i><sub>sat</sub> for a broad range of intrinsically disordered and multidomain proteins, including disease-associated RNA- and DNA-binding proteins involved in the formation of stress granules and P granules, as well as engineered mutants of hnRNPA1. Furthermore, we compare our TI methods against a computationally efficient machine-learning predictor trained to estimate saturation concentrations at room temperature. While both approaches yield realistic predictions, explicit molecular dynamics simulations enable the calculation of complete phase diagrams and provide insight into the molecular mechanisms and interactions driving phase separation. Overall, our approach offers a robust, physically grounded framework for improving and validating coarse-grained models of biomolecular phase behavior, effectively bridging the gap between simulation and experiment.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9919–9934"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Predicting Saturation Concentrations of Phase-Separating Proteins via Thermodynamic Integration\",\"authors\":\"Eduardo Pedraza, , , Andres R. Tejedor, , , Alejandro Feito, , , Francisco Gámez, , , Rosana Collepardo-Guevara, , , Eduardo Sanz*, , and , Jorge R. Espinosa*, \",\"doi\":\"10.1021/acs.jctc.5c00765\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Phase separation of proteins and nucleic acids into biomolecular condensates contributes to the regulation of cellular compartmentalization in membrane-less environments. A key parameter controlling the onset of biomolecular condensate formation is the saturation concentration (<i>C</i><sub>sat</sub>)─the threshold concentration above which condensation takes place. While measuring <i>C</i><sub>sat</sub> for protein solutions in vitro is experimentally accessible, determining this quantity in simulations remains challenging due to the extremely low equilibrium concentrations at which many proteins phase separate. This occurs because the gold standard in simulations consists of combining a residue-resolution coarse-grained model with the Direct Coexistence simulation method, which yields poor estimates of the equilibrium concentrations of the dilute phase due to lack of statistics. In this work, we present two independent thermodynamic integration (TI) schemes which, when combined with Direct Coexistence simulations, enable accurate calculation of saturation concentrations and phase diagrams─facilitating direct comparison with experimental measurements across a wide range of conditions. Our methods, combined with the Mpipi-Recharged residue-resolution model, accurately estimate <i>C</i><sub>sat</sub> for a broad range of intrinsically disordered and multidomain proteins, including disease-associated RNA- and DNA-binding proteins involved in the formation of stress granules and P granules, as well as engineered mutants of hnRNPA1. Furthermore, we compare our TI methods against a computationally efficient machine-learning predictor trained to estimate saturation concentrations at room temperature. While both approaches yield realistic predictions, explicit molecular dynamics simulations enable the calculation of complete phase diagrams and provide insight into the molecular mechanisms and interactions driving phase separation. Overall, our approach offers a robust, physically grounded framework for improving and validating coarse-grained models of biomolecular phase behavior, effectively bridging the gap between simulation and experiment.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"21 19\",\"pages\":\"9919–9934\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-09-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00765\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00765","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Predicting Saturation Concentrations of Phase-Separating Proteins via Thermodynamic Integration
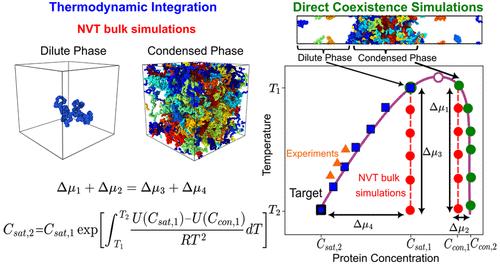
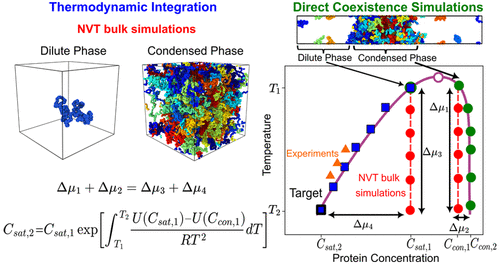
Phase separation of proteins and nucleic acids into biomolecular condensates contributes to the regulation of cellular compartmentalization in membrane-less environments. A key parameter controlling the onset of biomolecular condensate formation is the saturation concentration (Csat)─the threshold concentration above which condensation takes place. While measuring Csat for protein solutions in vitro is experimentally accessible, determining this quantity in simulations remains challenging due to the extremely low equilibrium concentrations at which many proteins phase separate. This occurs because the gold standard in simulations consists of combining a residue-resolution coarse-grained model with the Direct Coexistence simulation method, which yields poor estimates of the equilibrium concentrations of the dilute phase due to lack of statistics. In this work, we present two independent thermodynamic integration (TI) schemes which, when combined with Direct Coexistence simulations, enable accurate calculation of saturation concentrations and phase diagrams─facilitating direct comparison with experimental measurements across a wide range of conditions. Our methods, combined with the Mpipi-Recharged residue-resolution model, accurately estimate Csat for a broad range of intrinsically disordered and multidomain proteins, including disease-associated RNA- and DNA-binding proteins involved in the formation of stress granules and P granules, as well as engineered mutants of hnRNPA1. Furthermore, we compare our TI methods against a computationally efficient machine-learning predictor trained to estimate saturation concentrations at room temperature. While both approaches yield realistic predictions, explicit molecular dynamics simulations enable the calculation of complete phase diagrams and provide insight into the molecular mechanisms and interactions driving phase separation. Overall, our approach offers a robust, physically grounded framework for improving and validating coarse-grained models of biomolecular phase behavior, effectively bridging the gap between simulation and experiment.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


