Edoardo Spadetto*, , , Pier Herman Theodoor Philipsen*, , , Arno Förster*, , and , Lucas Visscher*,
{"title":"随机相位逼近的数值原子轨道和对偶互易空间网格的周期性实现。","authors":"Edoardo Spadetto*, , , Pier Herman Theodoor Philipsen*, , , Arno Förster*, , and , Lucas Visscher*, ","doi":"10.1021/acs.jctc.5c00751","DOIUrl":null,"url":null,"abstract":"<p >The random phase approximation (RPA) has emerged as a prominent first-principles method in material science, particularly to study the adsorption and chemisorption of small molecules on surfaces. However, its widespread application is hampered by its relatively high computational cost. Here, we present a well-parallelised implementation of the RPA with localized atomic orbitals and pair-atomic density fitting, which is especially suitable for studying two-dimensional systems. Through a dual <b><i>k</i></b>-grid scheme, we achieve fast and reliable convergence of RPA correlation energies to the thermodynamic limit. We demonstrate the efficacy of our implementation through an application to the adsorption of CO on MgO(001) using PBE input orbitals (RPA@PBE). Our calculated adsorption energy is in excellent agreement with previously published RPA@PBE studies, but, as expected, overestimates the experimentally available adsorption energies as well as recent CCSD(T) results.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9347–9363"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/pdf/10.1021/acs.jctc.5c00751","citationCount":"0","resultStr":"{\"title\":\"Periodic Implementation of the Random Phase Approximation with Numerical Atomic Orbitals and Dual Reciprocal Space Grids\",\"authors\":\"Edoardo Spadetto*, , , Pier Herman Theodoor Philipsen*, , , Arno Förster*, , and , Lucas Visscher*, \",\"doi\":\"10.1021/acs.jctc.5c00751\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The random phase approximation (RPA) has emerged as a prominent first-principles method in material science, particularly to study the adsorption and chemisorption of small molecules on surfaces. However, its widespread application is hampered by its relatively high computational cost. Here, we present a well-parallelised implementation of the RPA with localized atomic orbitals and pair-atomic density fitting, which is especially suitable for studying two-dimensional systems. Through a dual <b><i>k</i></b>-grid scheme, we achieve fast and reliable convergence of RPA correlation energies to the thermodynamic limit. We demonstrate the efficacy of our implementation through an application to the adsorption of CO on MgO(001) using PBE input orbitals (RPA@PBE). Our calculated adsorption energy is in excellent agreement with previously published RPA@PBE studies, but, as expected, overestimates the experimentally available adsorption energies as well as recent CCSD(T) results.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"21 19\",\"pages\":\"9347–9363\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-09-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/pdf/10.1021/acs.jctc.5c00751\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00751\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00751","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
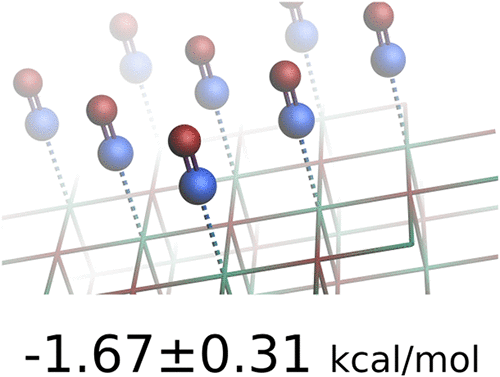
Periodic Implementation of the Random Phase Approximation with Numerical Atomic Orbitals and Dual Reciprocal Space Grids
The random phase approximation (RPA) has emerged as a prominent first-principles method in material science, particularly to study the adsorption and chemisorption of small molecules on surfaces. However, its widespread application is hampered by its relatively high computational cost. Here, we present a well-parallelised implementation of the RPA with localized atomic orbitals and pair-atomic density fitting, which is especially suitable for studying two-dimensional systems. Through a dual k-grid scheme, we achieve fast and reliable convergence of RPA correlation energies to the thermodynamic limit. We demonstrate the efficacy of our implementation through an application to the adsorption of CO on MgO(001) using PBE input orbitals (RPA@PBE). Our calculated adsorption energy is in excellent agreement with previously published RPA@PBE studies, but, as expected, overestimates the experimentally available adsorption energies as well as recent CCSD(T) results.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


