Vaishnavi A. Khalas, , , Satyam Shinde*, , and , Prahlad Baruah,
{"title":"综合DFT和实验方法研究牛皮素及其衍生物的结构、电子和光谱性质。","authors":"Vaishnavi A. Khalas, , , Satyam Shinde*, , and , Prahlad Baruah, ","doi":"10.1021/acs.jpca.5c04915","DOIUrl":null,"url":null,"abstract":"<p >Nobiletin, a polymethoxylated flavonoid with known antioxidant, anti-inflammatory, and anticancer potentials, is investigated, along with three derivatives, using an integrated experimental and computational approach. Density Functional Theory (DFT) is employed to evaluate the structural, electronic, and vibrational spectroscopic properties. To determine the most suitable computational conditions, we compared three functionals─B3LYP, M06–2X, and PBE0 using basis sets 6–31++G(d,p), def2-TZVP, and cc-PVDZ. The B3LYP/6–31++G(d,p) level was identified as the most reliable in terms of accuracy and computational efficiency and was subsequently used in combination with experimental infrared and Raman spectroscopic analyses. Vibrational mode assignments are carried out using the Potential Energy Distribution analysis, and theoretical spectra are validated against experimental results. Solvent effects in water and dimethyl sulfoxide are examined by using the Integral Equation Formalism Polarizable Continuum Model to assess the environmental influences on molecular properties. Frontier molecular orbital analysis suggests that the demethylated derivatives exhibit enhanced electronic behavior, including increased electron affinity, ionization, and electronegativity, indicating greater potential for molecular interaction. Potential (MESP) mapping identifies C═O and hydroxyl-substituted regions as key reactive sites susceptible to nucleophilic and electrophilic attack, respectively. Furthermore, a DFT guided derivatization scheme is proposed to generate NM1–NM3 from the parent compound with intermediate states evaluated through Gibbs free energy calculations to confirm thermodynamic feasibility. The findings offer comprehensive insights into the physicochemical behavior of Nobiletin and its derivatives and support their potential utility in anticancer drug development and biophysical research.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"129 41","pages":"9548–9558"},"PeriodicalIF":2.8000,"publicationDate":"2025-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"An Integrated DFT and Experimental Approach to Investigate the Structural, Electronic, and Spectroscopic Properties of Nobiletin and Its Derivatives\",\"authors\":\"Vaishnavi A. Khalas, , , Satyam Shinde*, , and , Prahlad Baruah, \",\"doi\":\"10.1021/acs.jpca.5c04915\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Nobiletin, a polymethoxylated flavonoid with known antioxidant, anti-inflammatory, and anticancer potentials, is investigated, along with three derivatives, using an integrated experimental and computational approach. Density Functional Theory (DFT) is employed to evaluate the structural, electronic, and vibrational spectroscopic properties. To determine the most suitable computational conditions, we compared three functionals─B3LYP, M06–2X, and PBE0 using basis sets 6–31++G(d,p), def2-TZVP, and cc-PVDZ. The B3LYP/6–31++G(d,p) level was identified as the most reliable in terms of accuracy and computational efficiency and was subsequently used in combination with experimental infrared and Raman spectroscopic analyses. Vibrational mode assignments are carried out using the Potential Energy Distribution analysis, and theoretical spectra are validated against experimental results. Solvent effects in water and dimethyl sulfoxide are examined by using the Integral Equation Formalism Polarizable Continuum Model to assess the environmental influences on molecular properties. Frontier molecular orbital analysis suggests that the demethylated derivatives exhibit enhanced electronic behavior, including increased electron affinity, ionization, and electronegativity, indicating greater potential for molecular interaction. Potential (MESP) mapping identifies C═O and hydroxyl-substituted regions as key reactive sites susceptible to nucleophilic and electrophilic attack, respectively. Furthermore, a DFT guided derivatization scheme is proposed to generate NM1–NM3 from the parent compound with intermediate states evaluated through Gibbs free energy calculations to confirm thermodynamic feasibility. The findings offer comprehensive insights into the physicochemical behavior of Nobiletin and its derivatives and support their potential utility in anticancer drug development and biophysical research.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"129 41\",\"pages\":\"9548–9558\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-09-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.5c04915\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.5c04915","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
An Integrated DFT and Experimental Approach to Investigate the Structural, Electronic, and Spectroscopic Properties of Nobiletin and Its Derivatives
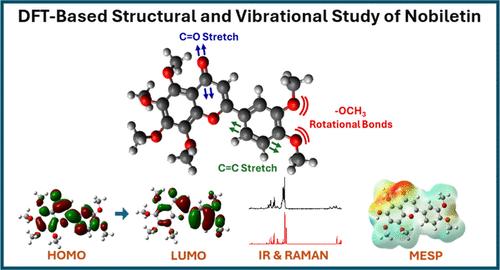
Nobiletin, a polymethoxylated flavonoid with known antioxidant, anti-inflammatory, and anticancer potentials, is investigated, along with three derivatives, using an integrated experimental and computational approach. Density Functional Theory (DFT) is employed to evaluate the structural, electronic, and vibrational spectroscopic properties. To determine the most suitable computational conditions, we compared three functionals─B3LYP, M06–2X, and PBE0 using basis sets 6–31++G(d,p), def2-TZVP, and cc-PVDZ. The B3LYP/6–31++G(d,p) level was identified as the most reliable in terms of accuracy and computational efficiency and was subsequently used in combination with experimental infrared and Raman spectroscopic analyses. Vibrational mode assignments are carried out using the Potential Energy Distribution analysis, and theoretical spectra are validated against experimental results. Solvent effects in water and dimethyl sulfoxide are examined by using the Integral Equation Formalism Polarizable Continuum Model to assess the environmental influences on molecular properties. Frontier molecular orbital analysis suggests that the demethylated derivatives exhibit enhanced electronic behavior, including increased electron affinity, ionization, and electronegativity, indicating greater potential for molecular interaction. Potential (MESP) mapping identifies C═O and hydroxyl-substituted regions as key reactive sites susceptible to nucleophilic and electrophilic attack, respectively. Furthermore, a DFT guided derivatization scheme is proposed to generate NM1–NM3 from the parent compound with intermediate states evaluated through Gibbs free energy calculations to confirm thermodynamic feasibility. The findings offer comprehensive insights into the physicochemical behavior of Nobiletin and its derivatives and support their potential utility in anticancer drug development and biophysical research.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


