{"title":"用从头算分子动力学揭示非水锂氧电池中Co-N-C单原子催化剂上电解质对氧还原的影响","authors":"Silan Chen, , , Qinghan Yu, , , Yujin Ji*, , and , Youyong Li*, ","doi":"10.1021/acscatal.5c03920","DOIUrl":null,"url":null,"abstract":"<p >Nonaqueous lithium–oxygen batteries (LOBs) hold immense promise due to their ultrahigh theoretical energy density, yet the role of electrolytes in regulating reaction kinetics during the oxygen reduction reaction (ORR) remains fundamentally unclear. Here, we systematically explore ORR pathways at the interface between Co–N–C single-atom catalysts (SACs) and electrolytes via <i>ab initio</i> molecular dynamics (AIMD). In bulk electrolytes, simulations reveal Li<sup>+</sup>-solvent bonding strength follows donor number (DN) order, which may result in a difference in the free energy of interfacial reaction. High-DN solvents elevate Li<sup>+</sup> insertion barrier due to strong Li<sup>+</sup>-solvent binding, while facilitating *LiO<sub>2</sub> desorption at the interface. However, the desorption of *LiO<sub>2</sub> into the electrolytes is found to be thermodynamically unfavorable, thereby driving the reaction toward surface-mediated growth. Low-DN electrolytes paired with high-adsorption catalysts enforce surface growth, while high-DN systems with weak-adsorption catalysts favor solution growth. Our work proposes a catalyst–electrolyte interfacial Li<sup>+</sup> competition principle that governs discharge product formation pathways and offers optimization strategies for LOBs.</p>","PeriodicalId":9,"journal":{"name":"ACS Catalysis ","volume":"15 19","pages":"16782–16791"},"PeriodicalIF":13.1000,"publicationDate":"2025-09-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Unveiling Electrolyte Effects on the Oxygen Reduction at Co–N–C Single-Atom Catalysts in Nonaqueous Lithium–Oxygen Batteries by Ab Initio Molecular Dynamics\",\"authors\":\"Silan Chen, , , Qinghan Yu, , , Yujin Ji*, , and , Youyong Li*, \",\"doi\":\"10.1021/acscatal.5c03920\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Nonaqueous lithium–oxygen batteries (LOBs) hold immense promise due to their ultrahigh theoretical energy density, yet the role of electrolytes in regulating reaction kinetics during the oxygen reduction reaction (ORR) remains fundamentally unclear. Here, we systematically explore ORR pathways at the interface between Co–N–C single-atom catalysts (SACs) and electrolytes via <i>ab initio</i> molecular dynamics (AIMD). In bulk electrolytes, simulations reveal Li<sup>+</sup>-solvent bonding strength follows donor number (DN) order, which may result in a difference in the free energy of interfacial reaction. High-DN solvents elevate Li<sup>+</sup> insertion barrier due to strong Li<sup>+</sup>-solvent binding, while facilitating *LiO<sub>2</sub> desorption at the interface. However, the desorption of *LiO<sub>2</sub> into the electrolytes is found to be thermodynamically unfavorable, thereby driving the reaction toward surface-mediated growth. Low-DN electrolytes paired with high-adsorption catalysts enforce surface growth, while high-DN systems with weak-adsorption catalysts favor solution growth. Our work proposes a catalyst–electrolyte interfacial Li<sup>+</sup> competition principle that governs discharge product formation pathways and offers optimization strategies for LOBs.</p>\",\"PeriodicalId\":9,\"journal\":{\"name\":\"ACS Catalysis \",\"volume\":\"15 19\",\"pages\":\"16782–16791\"},\"PeriodicalIF\":13.1000,\"publicationDate\":\"2025-09-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS Catalysis \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acscatal.5c03920\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Catalysis ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acscatal.5c03920","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Unveiling Electrolyte Effects on the Oxygen Reduction at Co–N–C Single-Atom Catalysts in Nonaqueous Lithium–Oxygen Batteries by Ab Initio Molecular Dynamics
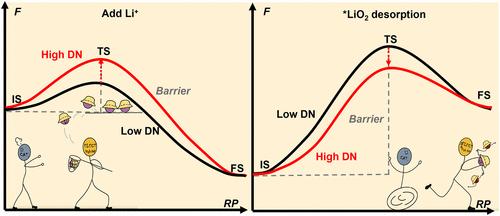
Nonaqueous lithium–oxygen batteries (LOBs) hold immense promise due to their ultrahigh theoretical energy density, yet the role of electrolytes in regulating reaction kinetics during the oxygen reduction reaction (ORR) remains fundamentally unclear. Here, we systematically explore ORR pathways at the interface between Co–N–C single-atom catalysts (SACs) and electrolytes via ab initio molecular dynamics (AIMD). In bulk electrolytes, simulations reveal Li+-solvent bonding strength follows donor number (DN) order, which may result in a difference in the free energy of interfacial reaction. High-DN solvents elevate Li+ insertion barrier due to strong Li+-solvent binding, while facilitating *LiO2 desorption at the interface. However, the desorption of *LiO2 into the electrolytes is found to be thermodynamically unfavorable, thereby driving the reaction toward surface-mediated growth. Low-DN electrolytes paired with high-adsorption catalysts enforce surface growth, while high-DN systems with weak-adsorption catalysts favor solution growth. Our work proposes a catalyst–electrolyte interfacial Li+ competition principle that governs discharge product formation pathways and offers optimization strategies for LOBs.
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


