{"title":"线粒体氧化应激、细胞损伤和干细胞衰老在复合体II电子传递缺陷早衰模型中的作用。","authors":"Takamasa Ishii, Kayo Yasuda, Masaki Miyazawa, Hiromi Onouchi, Sumino Yanase, Naoaki Ishii","doi":"10.3164/jcbn.25-62","DOIUrl":null,"url":null,"abstract":"<p><p>Mitochondria which are the major intracellular reactive oxygen species (ROS) sources produce especially superoxide anion (O<sub>2</sub> <sup>•-</sup>) as a byproduct of energy production. It has been well known that O<sub>2</sub> <sup>•-</sup> is converted from oxygen (O<sub>2</sub>) and is overproduced by excessive electron leakage from the mitochondrial electron transport chain (ETC), mainly complexes I and III. However we have previously reported that several point mutations (specifically G71E in <i>C. elegans</i>, I71E in <i>Drosophila</i> and V69E in mouse) in succinate dehydrogenase C subunit (SDHC) of complex II cause mitochondrial electron transport defect leading to O<sub>2</sub> <sup>•-</sup> overproduction from mitochondria. These mutations can cause endogenous oxidative stress resulting in tumorigenesis and apoptosis as well as premature death. Recently, we have also demonstrated that premature aging of hematopoietic stem cell with a mutation in SDHC is developed after the growth phase and normal development. Here, we review cellular damages by complex II electron transport defect-induced endogenous oxidative stress in premature aging models.</p>","PeriodicalId":15429,"journal":{"name":"Journal of Clinical Biochemistry and Nutrition","volume":"77 2","pages":"101-112"},"PeriodicalIF":1.7000,"publicationDate":"2025-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12440677/pdf/","citationCount":"0","resultStr":"{\"title\":\"Mitochondrial oxidative stress, cellular damages and stem cell aging in premature aging models with complex II electron transport defect.\",\"authors\":\"Takamasa Ishii, Kayo Yasuda, Masaki Miyazawa, Hiromi Onouchi, Sumino Yanase, Naoaki Ishii\",\"doi\":\"10.3164/jcbn.25-62\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Mitochondria which are the major intracellular reactive oxygen species (ROS) sources produce especially superoxide anion (O<sub>2</sub> <sup>•-</sup>) as a byproduct of energy production. It has been well known that O<sub>2</sub> <sup>•-</sup> is converted from oxygen (O<sub>2</sub>) and is overproduced by excessive electron leakage from the mitochondrial electron transport chain (ETC), mainly complexes I and III. However we have previously reported that several point mutations (specifically G71E in <i>C. elegans</i>, I71E in <i>Drosophila</i> and V69E in mouse) in succinate dehydrogenase C subunit (SDHC) of complex II cause mitochondrial electron transport defect leading to O<sub>2</sub> <sup>•-</sup> overproduction from mitochondria. These mutations can cause endogenous oxidative stress resulting in tumorigenesis and apoptosis as well as premature death. Recently, we have also demonstrated that premature aging of hematopoietic stem cell with a mutation in SDHC is developed after the growth phase and normal development. Here, we review cellular damages by complex II electron transport defect-induced endogenous oxidative stress in premature aging models.</p>\",\"PeriodicalId\":15429,\"journal\":{\"name\":\"Journal of Clinical Biochemistry and Nutrition\",\"volume\":\"77 2\",\"pages\":\"101-112\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2025-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12440677/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Clinical Biochemistry and Nutrition\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.3164/jcbn.25-62\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/7/2 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"NUTRITION & DIETETICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Biochemistry and Nutrition","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.3164/jcbn.25-62","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/7/2 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"NUTRITION & DIETETICS","Score":null,"Total":0}
Mitochondrial oxidative stress, cellular damages and stem cell aging in premature aging models with complex II electron transport defect.
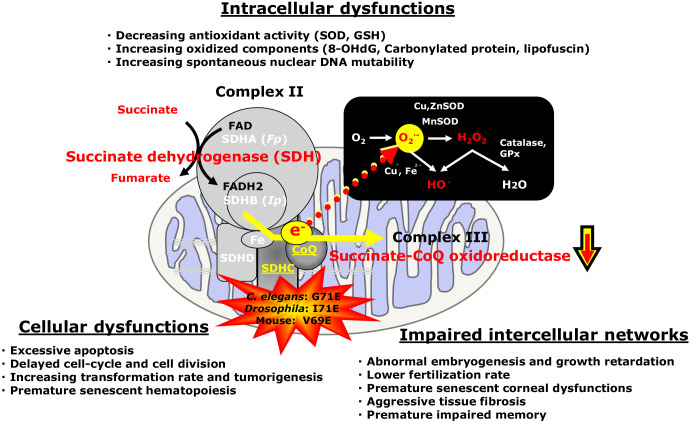
Mitochondria which are the major intracellular reactive oxygen species (ROS) sources produce especially superoxide anion (O2•-) as a byproduct of energy production. It has been well known that O2•- is converted from oxygen (O2) and is overproduced by excessive electron leakage from the mitochondrial electron transport chain (ETC), mainly complexes I and III. However we have previously reported that several point mutations (specifically G71E in C. elegans, I71E in Drosophila and V69E in mouse) in succinate dehydrogenase C subunit (SDHC) of complex II cause mitochondrial electron transport defect leading to O2•- overproduction from mitochondria. These mutations can cause endogenous oxidative stress resulting in tumorigenesis and apoptosis as well as premature death. Recently, we have also demonstrated that premature aging of hematopoietic stem cell with a mutation in SDHC is developed after the growth phase and normal development. Here, we review cellular damages by complex II electron transport defect-induced endogenous oxidative stress in premature aging models.
期刊介绍:
Journal of Clinical Biochemistry and Nutrition (JCBN) is
an international, interdisciplinary publication encompassing
chemical, biochemical, physiological, pathological, toxicological and medical approaches to research on lipid peroxidation, free radicals, oxidative stress and nutrition. The
Journal welcomes original contributions dealing with all
aspects of clinical biochemistry and clinical nutrition
including both in vitro and in vivo studies.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


