PyaiVS统一人工智能工作流程,加速配体发现并产生ABCG2抑制剂
IF 5.9
2区 医学
Q1 CHEMISTRY, MEDICINAL
引用次数: 0
摘要
开发用于虚拟筛选的优化人工智能模型需要协调选择算法、分子表示和数据分割策略,但缺乏集成工具。我们介绍PyaiVS,一个Python包,集成了九种机器学习算法,五种分子表示和三种数据分割策略。本研究表明,构建高效的人工智能驱动的小分子虚拟筛选模型需要协调优化算法架构(例如,优先考虑深度学习模型,如GCN、GAT和专心FP)、分子表征(小数据集的ECFP4/MACCS指纹和大规模数据的基于分子图的表征)和数据分割策略(基于聚类的分割实现68.5%最优AUC-ROC性能)。为了证明其实用性,我们将PyaiVS与药效团建模和对接相结合,筛选了4,188,623种ABCG2抑制剂化合物。通过实验验证,发现4个结合ABCG2的化合物(C1/C6/C7/C9) Kd值低于100 μM (5.31-51.35 μM),可增强拓扑替康的细胞毒性。PyaiVS通过将关键组件统一到一个可访问的平台来简化虚拟筛选,该平台可在https://github.com/danqingmk/OpenVS_PyaiVS免费获得。本文章由计算机程序翻译,如有差异,请以英文原文为准。
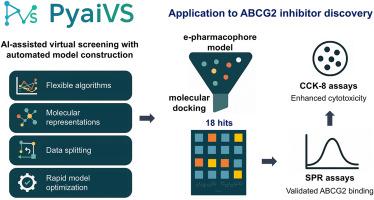
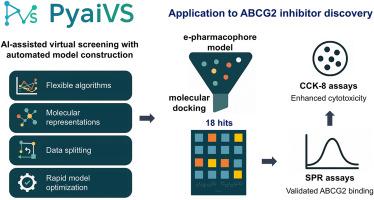
PyaiVS unifies AI workflows to accelerate ligand discovery and yields ABCG2 inhibitors
Developing optimized AI models for virtual screening requires coordinated selection of algorithms, molecular representations, and data splitting strategies, yet lacks integrated tools. We present PyaiVS, a Python package that integrates nine machine learning algorithms, five molecular representations, and three data splitting strategies. This study demonstrates that constructing efficient AI-driven virtual screening models for small molecules requires coordinated optimization of algorithm architectures (e.g., prioritizing deep learning models such as GCN, GAT, and Attentive FP), molecular representations (ECFP4/MACCS fingerprints for small datasets and molecular graph-based representations for large-scale data), and data splitting strategies (clustering-based splitting achieving 68.5 % optimal AUC-ROC performance). To demonstrate utility, we combined PyaiVS with pharmacophore modeling and docking to screen 4,188,623 compounds for ABCG2 inhibitors. Experimental validation identified four compounds (C1/C6/C7/C9) binding ABCG2 with sub-100 μM kd values (5.31–51.35 μM) that potentiate topotecan cytotoxicity. PyaiVS streamlines virtual screening by unifying critical components into an accessible platform, freely available at https://github.com/danqingmk/OpenVS_PyaiVS.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
11.70
自引率
9.00%
发文量
863
审稿时长
29 days
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


