{"title":"酮生成与酮分解缺陷的比较:30例患者回顾性单中心研究。","authors":"Ayca Burcu Kahraman, Yılmaz Yıldız, Ali Dursun, Serap Sivri, Turgay Coşkun, Ayşegül Tokatlı","doi":"10.5152/TurkArchPediatr.2025.25156","DOIUrl":null,"url":null,"abstract":"<p><p>Objective: Despite overlapping features, these 2 groups of disorders may exhibit distinct clinical and biochemical profiles. This study aimed to evaluate and compare the clinical presentation, laboratory findings, neuroimaging characteristics, genotypic spectrum, and clinical outcomes of patients with ketogenesis and ketolysis defects. Materials and Methods: Thirty patients diagnosed between 1986 and 2023 were retrospectively reviewed. Diagnosis was confirmed by clinical findings, biochemical, and genetic/enzymatic testing. Data included demographic details, clinical manifestations, neurodevelopmental status, laboratory results, imaging findings, genetic information, and treatments. Results: Of the 30 patients, 13 (43.3%) were diagnosed with 3-hydroxy-3-methylglutaryl-CoA lyase deficiency (HMGCLD), 14 (46.7%) with 2-methylacetoacetyl-coenzyme A thiolase deficiency (MATD), and 3 (10%) with succinyl-CoA:3-ketoacid CoA transferase deficiency (SCOTD). Patients with ketolysis defects presented at a later median age (210 vs. 30 days, P < .009) and exhibited more profound metabolic acidosis (pH 7.06 ± 0.18 vs. 7.26 ± 0.12, P = .028). Common presenting symptoms included vomiting in 25 (83.3%), hypoglycemia in 9 (33.3%), and seizures in 5 (16.6%). Leigh-like neuroimaging findings were observed in 3 HMGCLD patients. Biallelic pathogenic variants in HMGCL, ACAT1, or OXCT1 were identified in 14 patients. Dialysis was required in 1 MATD and 1 SCOTD case. Excluding those lost to follow-up, the mortality rates among the remaining 18 patients were 1/8, 12.5% in 2/9 HMGCLD, and 22.2% in MATD. One of the patients with SCOTD was alive at the time of the last follow-up. Conclusion: Patients with ketolysis defects are more likely to present later and with severe metabolic acidosis, occasionally requiring renal replacement therapy. Delayed diagnosis may hinder timely intervention, potentially contributing to increased mortality.</p>","PeriodicalId":75267,"journal":{"name":"Turkish archives of pediatrics","volume":"60 5","pages":"491-499"},"PeriodicalIF":1.7000,"publicationDate":"2025-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12432195/pdf/","citationCount":"0","resultStr":"{\"title\":\"Comparison of Ketogenesis and Ketolysis Defects: A Retrospective Single-Center Study of 30 Patients.\",\"authors\":\"Ayca Burcu Kahraman, Yılmaz Yıldız, Ali Dursun, Serap Sivri, Turgay Coşkun, Ayşegül Tokatlı\",\"doi\":\"10.5152/TurkArchPediatr.2025.25156\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Objective: Despite overlapping features, these 2 groups of disorders may exhibit distinct clinical and biochemical profiles. This study aimed to evaluate and compare the clinical presentation, laboratory findings, neuroimaging characteristics, genotypic spectrum, and clinical outcomes of patients with ketogenesis and ketolysis defects. Materials and Methods: Thirty patients diagnosed between 1986 and 2023 were retrospectively reviewed. Diagnosis was confirmed by clinical findings, biochemical, and genetic/enzymatic testing. Data included demographic details, clinical manifestations, neurodevelopmental status, laboratory results, imaging findings, genetic information, and treatments. Results: Of the 30 patients, 13 (43.3%) were diagnosed with 3-hydroxy-3-methylglutaryl-CoA lyase deficiency (HMGCLD), 14 (46.7%) with 2-methylacetoacetyl-coenzyme A thiolase deficiency (MATD), and 3 (10%) with succinyl-CoA:3-ketoacid CoA transferase deficiency (SCOTD). Patients with ketolysis defects presented at a later median age (210 vs. 30 days, P < .009) and exhibited more profound metabolic acidosis (pH 7.06 ± 0.18 vs. 7.26 ± 0.12, P = .028). Common presenting symptoms included vomiting in 25 (83.3%), hypoglycemia in 9 (33.3%), and seizures in 5 (16.6%). Leigh-like neuroimaging findings were observed in 3 HMGCLD patients. Biallelic pathogenic variants in HMGCL, ACAT1, or OXCT1 were identified in 14 patients. Dialysis was required in 1 MATD and 1 SCOTD case. Excluding those lost to follow-up, the mortality rates among the remaining 18 patients were 1/8, 12.5% in 2/9 HMGCLD, and 22.2% in MATD. One of the patients with SCOTD was alive at the time of the last follow-up. Conclusion: Patients with ketolysis defects are more likely to present later and with severe metabolic acidosis, occasionally requiring renal replacement therapy. Delayed diagnosis may hinder timely intervention, potentially contributing to increased mortality.</p>\",\"PeriodicalId\":75267,\"journal\":{\"name\":\"Turkish archives of pediatrics\",\"volume\":\"60 5\",\"pages\":\"491-499\"},\"PeriodicalIF\":1.7000,\"publicationDate\":\"2025-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12432195/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Turkish archives of pediatrics\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.5152/TurkArchPediatr.2025.25156\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"PEDIATRICS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Turkish archives of pediatrics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5152/TurkArchPediatr.2025.25156","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"PEDIATRICS","Score":null,"Total":0}
引用次数: 0
摘要
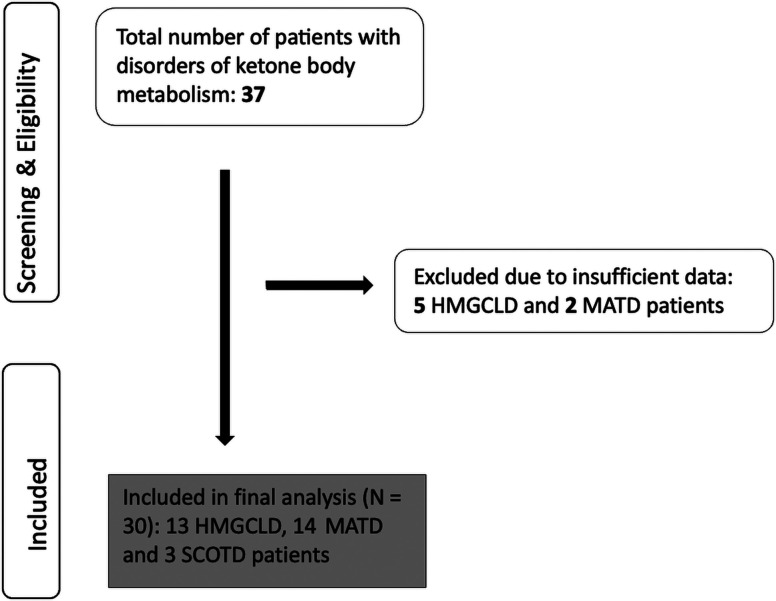
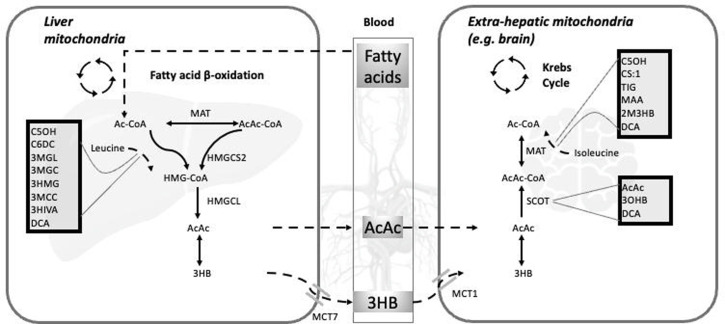
目的:尽管有重叠的特征,这两组疾病可能表现出不同的临床和生化特征。本研究旨在评估和比较生酮和解酮缺陷患者的临床表现、实验室结果、神经影像学特征、基因型谱和临床结果。材料和方法:回顾性分析1986年至2023年间诊断的30例患者。诊断由临床表现、生化和基因/酶检测证实。数据包括人口学细节、临床表现、神经发育状况、实验室结果、影像学发现、遗传信息和治疗。结果:30例患者中,13例(43.3%)诊断为3-羟基-3-甲基戊二酰辅酶A裂解酶缺乏症(HMGCLD), 14例(46.7%)诊断为2-甲基乙酰乙酰辅酶A巯基酶缺乏症(MATD), 3例(10%)诊断为琥珀酰辅酶A:3-酮酸辅酶A转移酶缺乏症(SCOTD)。酮解缺陷患者出现的中位年龄较晚(210 vs 30天,P < 0.009),代谢性酸中毒更严重(pH值7.06±0.18 vs 7.26±0.12,P = 0.028)。常见的症状包括呕吐25例(83.3%),低血糖9例(33.3%),癫痫发作5例(16.6%)。3例HMGCLD患者行leigh样神经影像学检查。在14例患者中发现HMGCL、ACAT1或OXCT1的双等位致病变异。1例MATD和1例SCOTD需要透析。排除失访者,其余18例患者的死亡率为1/8,2/9 HMGCLD的死亡率为12.5%,MATD的死亡率为22.2%。在最后一次随访时,一名患有SCOTD的患者还活着。结论:有酮解缺陷的患者更容易出现较晚的代谢性酸中毒,偶尔需要肾脏替代治疗。延迟诊断可能妨碍及时干预,可能导致死亡率增加。
Comparison of Ketogenesis and Ketolysis Defects: A Retrospective Single-Center Study of 30 Patients.
Objective: Despite overlapping features, these 2 groups of disorders may exhibit distinct clinical and biochemical profiles. This study aimed to evaluate and compare the clinical presentation, laboratory findings, neuroimaging characteristics, genotypic spectrum, and clinical outcomes of patients with ketogenesis and ketolysis defects. Materials and Methods: Thirty patients diagnosed between 1986 and 2023 were retrospectively reviewed. Diagnosis was confirmed by clinical findings, biochemical, and genetic/enzymatic testing. Data included demographic details, clinical manifestations, neurodevelopmental status, laboratory results, imaging findings, genetic information, and treatments. Results: Of the 30 patients, 13 (43.3%) were diagnosed with 3-hydroxy-3-methylglutaryl-CoA lyase deficiency (HMGCLD), 14 (46.7%) with 2-methylacetoacetyl-coenzyme A thiolase deficiency (MATD), and 3 (10%) with succinyl-CoA:3-ketoacid CoA transferase deficiency (SCOTD). Patients with ketolysis defects presented at a later median age (210 vs. 30 days, P < .009) and exhibited more profound metabolic acidosis (pH 7.06 ± 0.18 vs. 7.26 ± 0.12, P = .028). Common presenting symptoms included vomiting in 25 (83.3%), hypoglycemia in 9 (33.3%), and seizures in 5 (16.6%). Leigh-like neuroimaging findings were observed in 3 HMGCLD patients. Biallelic pathogenic variants in HMGCL, ACAT1, or OXCT1 were identified in 14 patients. Dialysis was required in 1 MATD and 1 SCOTD case. Excluding those lost to follow-up, the mortality rates among the remaining 18 patients were 1/8, 12.5% in 2/9 HMGCLD, and 22.2% in MATD. One of the patients with SCOTD was alive at the time of the last follow-up. Conclusion: Patients with ketolysis defects are more likely to present later and with severe metabolic acidosis, occasionally requiring renal replacement therapy. Delayed diagnosis may hinder timely intervention, potentially contributing to increased mortality.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


