新型喹啉类磺胺吡啶衍生物PI3K/HDAC双靶点抑制剂的设计、合成及生物活性评价
IF 5.9
2区 医学
Q1 CHEMISTRY, MEDICINAL
引用次数: 0
摘要
在恶性肿瘤的治疗中,双靶点抑制剂可有效避免单靶点给药引起的其他信号通路代偿性激活,以及共给药引起的药代动力学改变和患者依从性差。在这项研究中,我们选择了磷脂酰肌醇3-激酶(PI3K)和组蛋白去乙酰化酶(HDAC)这两个肿瘤细胞发育的关键靶点,设计了具有不同类型和长度的连锁链的双抑制剂。选择喹啉连接的磺胺吡啶片段作为PI3K抑制的药效团,选择邻氨基苯甲酰胺片段作为HDAC的Zn2+结合基团(ZBG)的药效团,是为了最大限度地提高这两个靶点的药代动力学特性。利用这些连锁链,我们设计了41种具有独特结构的新型喹啉连接磺胺吡啶PI3K/HDAC双靶点抑制剂。这些化合物大多对Jurkat、K562、MCF-7和PC9R细胞有较强的抗增殖作用。具体来说,SJY26不仅对肿瘤细胞表现出强大的抗增殖活性,而且对PI3Kα和HDAC1也表现出明显的抑制活性。此外,SJY26在1.25 μM下显著抑制PC9R细胞的迁移,降低AKT磷酸化,降低组蛋白H3去乙酰化。总之,本研究强调了新型喹啉连接的磺胺吡啶衍生物作为PI3K/HDAC双靶点抑制剂的治疗潜力,值得进一步研究。本文章由计算机程序翻译,如有差异,请以英文原文为准。
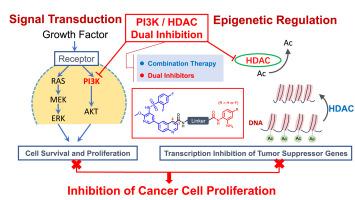
Design, synthesis and bioactive evaluation of novel quinoline-linked sulfonamide-pyridine derivatives as PI3K/HDAC dual-target inhibitors
In the treatment of malignant tumors, dual-target inhibitors can effectively avoid the compensatory activation of other signaling pathways caused by single-target administration, as well as the pharmacokinetic changes and poor patient compliance associated with co-administration. For this study, we selected phosphatidylinositol 3-kinase (PI3K) and histone deacetylase (HDAC), two critical targets in tumor cell development, and designed dual inhibitors displaying various types and lengths of linkage chains. A quinoline-linked sulfonamide-pyridine fragment as the pharmacophore for PI3K inhibition, and an o-aminobenzamide fragment as the pharmacophore for the Zn2+-binding group (ZBG) of HDAC have been chosen in order to maximize the pharmacokinetic properties of both targets. Using these linkage chains, we designed 41 novel quinoline-linked sulfonamide-pyridine PI3K/HDAC dual-target inhibitors with unique structures. Most of these compounds exhibited strong antiproliferative effects on Jurkat, K562, MCF-7, and PC9R cells. Specifically, SJY26 not only demonstrated potent antiproliferative activity against tumor cells but also exhibited outstanding inhibitory activity against PI3Kα and HDAC1. Moreover, SJY26 significantly inhibited the migration of PC9R cells at 1.25 μM, reduced AKT phosphorylation, and decreased histone H3 deacetylation. In summary, this research highlights the promising therapeutic potential of novel quinoline-linked sulfonamide-pyridine derivatives as PI3K/HDAC dual-target inhibitors, warranting further investigation.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

CiteScore
11.70
自引率
9.00%
发文量
863
审稿时长
29 days
期刊介绍:
The European Journal of Medicinal Chemistry is a global journal that publishes studies on all aspects of medicinal chemistry. It provides a medium for publication of original papers and also welcomes critical review papers.
A typical paper would report on the organic synthesis, characterization and pharmacological evaluation of compounds. Other topics of interest are drug design, QSAR, molecular modeling, drug-receptor interactions, molecular aspects of drug metabolism, prodrug synthesis and drug targeting. The journal expects manuscripts to present the rational for a study, provide insight into the design of compounds or understanding of mechanism, or clarify the targets.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


