{"title":"利用多状态重加权和配置空间映射优化的大压力和温度范围内具有改进热力学性质的水模型。","authors":"Himanshu Paliwal, and , Michael R. Shirts*, ","doi":"10.1021/acs.jctc.5c00978","DOIUrl":null,"url":null,"abstract":"<p >We show how reweighting and configuration mapping algorithms can be used to efficiently optimize molecular models using thermodynamic properties at a large number of state points from molecular simulations. As a proof of concept, we perform a multidimensional, multiobjective parameterization of a rigid water model over a large pressure [1–5000 atm] and temperature [274.15–372.15 K] range to the experimental property surfaces estimated using the IAPWS95 equation of state for water. Over 4000 parameter combinations in a six-dimensional parameter space were explored during the minimization. A similar parameterization with standard techniques would have taken more than 250 CPU years, but with the application of the newly developed techniques, the computational time was reduced to four CPU months. Without the added efficiency of the methods presented here, the optimization could not have simultaneously taken into account the large range of temperature and pressure points used in the fitting. The paper also describes how and why incorporating the thermodynamic properties from the first and second derivatives of Gibbs energy into the objective function helps improve the parameterization process. The resulting water model reproduces liquid phase density within the upper limit of experimental uncertainty of 0.02% over a large range of temperatures and pressures, the most accurate model yet at this low level of theory over the entire range of temperature and pressure, with little loss of fidelity in other properties. We compare the performance of this water model with 11 other rigid water models in predicting a number of other thermodynamic and kinetic properties. This process illustrates the surprising fact that a simple point charge model is able to accurately capture a substantial range of both temperature- and pressure-dependent thermodynamics without substantial deviation from experiment at ambient temperatures and pressures.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 19","pages":"9687–9709"},"PeriodicalIF":5.5000,"publicationDate":"2025-09-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"GOPAL: A Water Model with Improved Thermodynamic Properties over a Large Pressure and Temperature Range Optimized Using Multistate Reweighting and Configuration Space Mapping\",\"authors\":\"Himanshu Paliwal, and , Michael R. Shirts*, \",\"doi\":\"10.1021/acs.jctc.5c00978\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >We show how reweighting and configuration mapping algorithms can be used to efficiently optimize molecular models using thermodynamic properties at a large number of state points from molecular simulations. As a proof of concept, we perform a multidimensional, multiobjective parameterization of a rigid water model over a large pressure [1–5000 atm] and temperature [274.15–372.15 K] range to the experimental property surfaces estimated using the IAPWS95 equation of state for water. Over 4000 parameter combinations in a six-dimensional parameter space were explored during the minimization. A similar parameterization with standard techniques would have taken more than 250 CPU years, but with the application of the newly developed techniques, the computational time was reduced to four CPU months. Without the added efficiency of the methods presented here, the optimization could not have simultaneously taken into account the large range of temperature and pressure points used in the fitting. The paper also describes how and why incorporating the thermodynamic properties from the first and second derivatives of Gibbs energy into the objective function helps improve the parameterization process. The resulting water model reproduces liquid phase density within the upper limit of experimental uncertainty of 0.02% over a large range of temperatures and pressures, the most accurate model yet at this low level of theory over the entire range of temperature and pressure, with little loss of fidelity in other properties. We compare the performance of this water model with 11 other rigid water models in predicting a number of other thermodynamic and kinetic properties. This process illustrates the surprising fact that a simple point charge model is able to accurately capture a substantial range of both temperature- and pressure-dependent thermodynamics without substantial deviation from experiment at ambient temperatures and pressures.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"21 19\",\"pages\":\"9687–9709\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-09-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00978\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c00978","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
GOPAL: A Water Model with Improved Thermodynamic Properties over a Large Pressure and Temperature Range Optimized Using Multistate Reweighting and Configuration Space Mapping
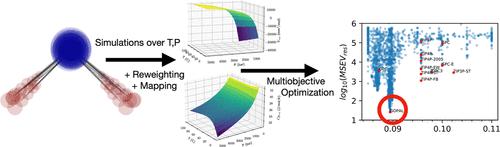
We show how reweighting and configuration mapping algorithms can be used to efficiently optimize molecular models using thermodynamic properties at a large number of state points from molecular simulations. As a proof of concept, we perform a multidimensional, multiobjective parameterization of a rigid water model over a large pressure [1–5000 atm] and temperature [274.15–372.15 K] range to the experimental property surfaces estimated using the IAPWS95 equation of state for water. Over 4000 parameter combinations in a six-dimensional parameter space were explored during the minimization. A similar parameterization with standard techniques would have taken more than 250 CPU years, but with the application of the newly developed techniques, the computational time was reduced to four CPU months. Without the added efficiency of the methods presented here, the optimization could not have simultaneously taken into account the large range of temperature and pressure points used in the fitting. The paper also describes how and why incorporating the thermodynamic properties from the first and second derivatives of Gibbs energy into the objective function helps improve the parameterization process. The resulting water model reproduces liquid phase density within the upper limit of experimental uncertainty of 0.02% over a large range of temperatures and pressures, the most accurate model yet at this low level of theory over the entire range of temperature and pressure, with little loss of fidelity in other properties. We compare the performance of this water model with 11 other rigid water models in predicting a number of other thermodynamic and kinetic properties. This process illustrates the surprising fact that a simple point charge model is able to accurately capture a substantial range of both temperature- and pressure-dependent thermodynamics without substantial deviation from experiment at ambient temperatures and pressures.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


