{"title":"CO2电还原Cu-Zn催化剂表面结构和活性景观的机器学习驱动映射","authors":"Yingru Wang, and , Liang Cao*, ","doi":"10.1021/acscatal.5c03337","DOIUrl":null,"url":null,"abstract":"<p >Electrocatalytic CO<sub>2</sub> reduction using bimetallic Cu-based catalysts offers a promising route for carbon-neutral carbon utilization. However, a lack of an atomic-scale understanding of active sites hinders the rational design of high-performance catalysts. In this work, we develop a machine-learning cluster expansion (CE) model, trained by density functional theory (DFT) calculations, to explore structure–activity relationships on Zn-doped Cu(111) surfaces for CO<sub>2</sub> to CO conversion. By incorporating a Bayesian machine learning approach with leave-one-out cross-validation into the CE model fitting, we achieve high predictive accuracy while lowering the overfitting risk, even with a relatively small training set. Metropolis Monte Carlo simulations based on the CE model predict thermodynamically stable surface configurations, *CO adsorption energies, and turnover frequencies (TOF) across a broad range of Zn compositions. Our results show that Zn into Cu(111) significantly enhances catalytic activity, with an optimal Zn doping level of ∼15%, yielding a TOF approximately 28 times higher than that of pure Cu(111). This enhancement results from Zn surface segregation and the formation of Cu active sites modulated by Zn coordination. Specifically, the number of neighboring Zn atoms, such as three or four first-nearest-neighbor (first-NN) Zn atoms, fine-tunes *CO adsorption energies on Cu, placing them within the optimal activity window. This work provides atomic-level insights into the role of the local alloy structure in catalytic performance and offers a generalizable strategy for active site engineering in bimetallic electrocatalysts.</p>","PeriodicalId":9,"journal":{"name":"ACS Catalysis ","volume":"15 19","pages":"16591–16599"},"PeriodicalIF":13.1000,"publicationDate":"2025-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Machine Learning–Driven Mapping of the Surface Structure and Activity Landscape in Cu–Zn Catalysts for CO2 Electroreduction\",\"authors\":\"Yingru Wang, and , Liang Cao*, \",\"doi\":\"10.1021/acscatal.5c03337\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Electrocatalytic CO<sub>2</sub> reduction using bimetallic Cu-based catalysts offers a promising route for carbon-neutral carbon utilization. However, a lack of an atomic-scale understanding of active sites hinders the rational design of high-performance catalysts. In this work, we develop a machine-learning cluster expansion (CE) model, trained by density functional theory (DFT) calculations, to explore structure–activity relationships on Zn-doped Cu(111) surfaces for CO<sub>2</sub> to CO conversion. By incorporating a Bayesian machine learning approach with leave-one-out cross-validation into the CE model fitting, we achieve high predictive accuracy while lowering the overfitting risk, even with a relatively small training set. Metropolis Monte Carlo simulations based on the CE model predict thermodynamically stable surface configurations, *CO adsorption energies, and turnover frequencies (TOF) across a broad range of Zn compositions. Our results show that Zn into Cu(111) significantly enhances catalytic activity, with an optimal Zn doping level of ∼15%, yielding a TOF approximately 28 times higher than that of pure Cu(111). This enhancement results from Zn surface segregation and the formation of Cu active sites modulated by Zn coordination. Specifically, the number of neighboring Zn atoms, such as three or four first-nearest-neighbor (first-NN) Zn atoms, fine-tunes *CO adsorption energies on Cu, placing them within the optimal activity window. This work provides atomic-level insights into the role of the local alloy structure in catalytic performance and offers a generalizable strategy for active site engineering in bimetallic electrocatalysts.</p>\",\"PeriodicalId\":9,\"journal\":{\"name\":\"ACS Catalysis \",\"volume\":\"15 19\",\"pages\":\"16591–16599\"},\"PeriodicalIF\":13.1000,\"publicationDate\":\"2025-09-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ACS Catalysis \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acscatal.5c03337\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ACS Catalysis ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acscatal.5c03337","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Machine Learning–Driven Mapping of the Surface Structure and Activity Landscape in Cu–Zn Catalysts for CO2 Electroreduction
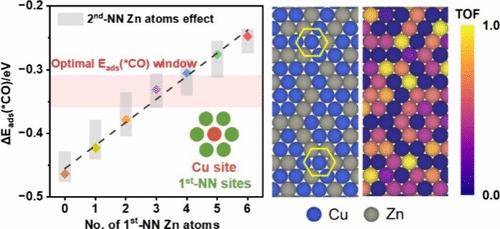
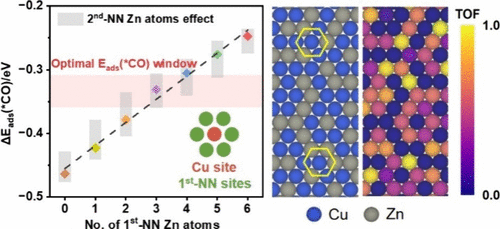
Electrocatalytic CO2 reduction using bimetallic Cu-based catalysts offers a promising route for carbon-neutral carbon utilization. However, a lack of an atomic-scale understanding of active sites hinders the rational design of high-performance catalysts. In this work, we develop a machine-learning cluster expansion (CE) model, trained by density functional theory (DFT) calculations, to explore structure–activity relationships on Zn-doped Cu(111) surfaces for CO2 to CO conversion. By incorporating a Bayesian machine learning approach with leave-one-out cross-validation into the CE model fitting, we achieve high predictive accuracy while lowering the overfitting risk, even with a relatively small training set. Metropolis Monte Carlo simulations based on the CE model predict thermodynamically stable surface configurations, *CO adsorption energies, and turnover frequencies (TOF) across a broad range of Zn compositions. Our results show that Zn into Cu(111) significantly enhances catalytic activity, with an optimal Zn doping level of ∼15%, yielding a TOF approximately 28 times higher than that of pure Cu(111). This enhancement results from Zn surface segregation and the formation of Cu active sites modulated by Zn coordination. Specifically, the number of neighboring Zn atoms, such as three or four first-nearest-neighbor (first-NN) Zn atoms, fine-tunes *CO adsorption energies on Cu, placing them within the optimal activity window. This work provides atomic-level insights into the role of the local alloy structure in catalytic performance and offers a generalizable strategy for active site engineering in bimetallic electrocatalysts.
期刊介绍:
ACS Catalysis is an esteemed journal that publishes original research in the fields of heterogeneous catalysis, molecular catalysis, and biocatalysis. It offers broad coverage across diverse areas such as life sciences, organometallics and synthesis, photochemistry and electrochemistry, drug discovery and synthesis, materials science, environmental protection, polymer discovery and synthesis, and energy and fuels.
The scope of the journal is to showcase innovative work in various aspects of catalysis. This includes new reactions and novel synthetic approaches utilizing known catalysts, the discovery or modification of new catalysts, elucidation of catalytic mechanisms through cutting-edge investigations, practical enhancements of existing processes, as well as conceptual advances in the field. Contributions to ACS Catalysis can encompass both experimental and theoretical research focused on catalytic molecules, macromolecules, and materials that exhibit catalytic turnover.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


