Davide Mitoli, , , Alessandro Erba, , , Vincenzo Barone, , and , Marco Mendolicchio*,
{"title":"分子晶体中的非调和性:广义微扰理论与周期计算","authors":"Davide Mitoli, , , Alessandro Erba, , , Vincenzo Barone, , and , Marco Mendolicchio*, ","doi":"10.1021/acs.jpclett.5c02217","DOIUrl":null,"url":null,"abstract":"<p >Accurate simulation of vibrational spectra in the solid state remains a major challenge due to the combined effects of anharmonicity, intermolecular interactions, and resonance phenomena. In this work, we introduce a generalized second-order vibrational perturbation theory (GVPT2) framework for the quantitative computational spectroscopy of molecular solids. The method balances efficiency and accuracy through a perturb-then-diagonalize approach in which resonant terms are excluded in the initial perturbative treatment and subsequently handled more accurately through a variational approach. This strategy ensures numerical stability while capturing essential vibrational couplings. As a representative application, we investigated the infrared spectrum of solid carbon dioxide (dry ice), a prototypical system exhibiting strong anharmonic effects and Fermi resonances. The generalized VPT2 approach accurately reproduces both absolute band positions and splitting patterns, yielding results in excellent agreement with the experimental data. These findings demonstrate the potential of the method for reliable and transferable anharmonic vibrational analysis across a broad class of solid-state systems.</p>","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"16 38","pages":"9956–9962"},"PeriodicalIF":4.6000,"publicationDate":"2025-09-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.acs.org/doi/pdf/10.1021/acs.jpclett.5c02217","citationCount":"0","resultStr":"{\"title\":\"Anharmonicity in Molecular Crystals: Generalized Perturbation Theory Meets Periodic Computations\",\"authors\":\"Davide Mitoli, , , Alessandro Erba, , , Vincenzo Barone, , and , Marco Mendolicchio*, \",\"doi\":\"10.1021/acs.jpclett.5c02217\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Accurate simulation of vibrational spectra in the solid state remains a major challenge due to the combined effects of anharmonicity, intermolecular interactions, and resonance phenomena. In this work, we introduce a generalized second-order vibrational perturbation theory (GVPT2) framework for the quantitative computational spectroscopy of molecular solids. The method balances efficiency and accuracy through a perturb-then-diagonalize approach in which resonant terms are excluded in the initial perturbative treatment and subsequently handled more accurately through a variational approach. This strategy ensures numerical stability while capturing essential vibrational couplings. As a representative application, we investigated the infrared spectrum of solid carbon dioxide (dry ice), a prototypical system exhibiting strong anharmonic effects and Fermi resonances. The generalized VPT2 approach accurately reproduces both absolute band positions and splitting patterns, yielding results in excellent agreement with the experimental data. These findings demonstrate the potential of the method for reliable and transferable anharmonic vibrational analysis across a broad class of solid-state systems.</p>\",\"PeriodicalId\":62,\"journal\":{\"name\":\"The Journal of Physical Chemistry Letters\",\"volume\":\"16 38\",\"pages\":\"9956–9962\"},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2025-09-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.acs.org/doi/pdf/10.1021/acs.jpclett.5c02217\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry Letters\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpclett.5c02217\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpclett.5c02217","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Anharmonicity in Molecular Crystals: Generalized Perturbation Theory Meets Periodic Computations
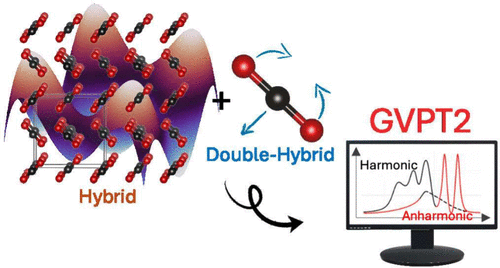
Accurate simulation of vibrational spectra in the solid state remains a major challenge due to the combined effects of anharmonicity, intermolecular interactions, and resonance phenomena. In this work, we introduce a generalized second-order vibrational perturbation theory (GVPT2) framework for the quantitative computational spectroscopy of molecular solids. The method balances efficiency and accuracy through a perturb-then-diagonalize approach in which resonant terms are excluded in the initial perturbative treatment and subsequently handled more accurately through a variational approach. This strategy ensures numerical stability while capturing essential vibrational couplings. As a representative application, we investigated the infrared spectrum of solid carbon dioxide (dry ice), a prototypical system exhibiting strong anharmonic effects and Fermi resonances. The generalized VPT2 approach accurately reproduces both absolute band positions and splitting patterns, yielding results in excellent agreement with the experimental data. These findings demonstrate the potential of the method for reliable and transferable anharmonic vibrational analysis across a broad class of solid-state systems.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


