Seyda Gul Ozcan, Eylul Koc, Ahmet Murt, Mevlut Tamer Dincer, Iclal Gurses, Nurhan Seyahi, Sinan Trabulus
{"title":"罕见的双重肾小球病理:Alport综合征和免疫复合物介导的MPGN。","authors":"Seyda Gul Ozcan, Eylul Koc, Ahmet Murt, Mevlut Tamer Dincer, Iclal Gurses, Nurhan Seyahi, Sinan Trabulus","doi":"10.1186/s12882-025-04400-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Immune complex-mediated membranoproliferative glomerulonephritis (IC-MPGN) and Alport syndrome are distinct glomerular diseases with different pathophysiologic mechanisms. Their coexistence is extremely rare and may present diagnostic and therapeutic challenges.</p><p><strong>Case presentation: </strong>A 42-year-old woman presented with persistent proteinuria and hematuria. Initial laboratory evaluations showed a progressive increase in proteinuria. She had a family history of renal disease. A renal biopsy, which had initially been postponed on patient request, subsequently revealed diffuse mesangial and focal endocapillary proliferation, GBM thickening, periglomerular fibrosis, and immune complex deposition with IgM, C3, and C1q, thereby confirming the diagnosis of IC-MPGN. Genetic testing identified a heterozygous COL4A5 c.1871G > A (p.Gly624Asp) mutation consistent with X-linked Alport syndrome. The patient follows conservative management now.</p><p><strong>Conclusion: </strong>This case highlights the rare coexistence of IC-MPGN, chronic TIN and Alport syndrome. In patients with progressive glomerular disease, especially with a family history of renal disease or extrarenal findings, genetic evaluation should be considered. Early renal biopsy and genetic testing are essential to guide management in complex cases with overlapping features.</p>","PeriodicalId":9089,"journal":{"name":"BMC Nephrology","volume":"26 1","pages":"514"},"PeriodicalIF":2.4000,"publicationDate":"2025-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12409941/pdf/","citationCount":"0","resultStr":"{\"title\":\"A rare case of dual glomerular pathology: Alport syndrome and immune complex-mediated MPGN.\",\"authors\":\"Seyda Gul Ozcan, Eylul Koc, Ahmet Murt, Mevlut Tamer Dincer, Iclal Gurses, Nurhan Seyahi, Sinan Trabulus\",\"doi\":\"10.1186/s12882-025-04400-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Immune complex-mediated membranoproliferative glomerulonephritis (IC-MPGN) and Alport syndrome are distinct glomerular diseases with different pathophysiologic mechanisms. Their coexistence is extremely rare and may present diagnostic and therapeutic challenges.</p><p><strong>Case presentation: </strong>A 42-year-old woman presented with persistent proteinuria and hematuria. Initial laboratory evaluations showed a progressive increase in proteinuria. She had a family history of renal disease. A renal biopsy, which had initially been postponed on patient request, subsequently revealed diffuse mesangial and focal endocapillary proliferation, GBM thickening, periglomerular fibrosis, and immune complex deposition with IgM, C3, and C1q, thereby confirming the diagnosis of IC-MPGN. Genetic testing identified a heterozygous COL4A5 c.1871G > A (p.Gly624Asp) mutation consistent with X-linked Alport syndrome. The patient follows conservative management now.</p><p><strong>Conclusion: </strong>This case highlights the rare coexistence of IC-MPGN, chronic TIN and Alport syndrome. In patients with progressive glomerular disease, especially with a family history of renal disease or extrarenal findings, genetic evaluation should be considered. Early renal biopsy and genetic testing are essential to guide management in complex cases with overlapping features.</p>\",\"PeriodicalId\":9089,\"journal\":{\"name\":\"BMC Nephrology\",\"volume\":\"26 1\",\"pages\":\"514\"},\"PeriodicalIF\":2.4000,\"publicationDate\":\"2025-09-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12409941/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Nephrology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s12882-025-04400-z\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"UROLOGY & NEPHROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Nephrology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12882-025-04400-z","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"UROLOGY & NEPHROLOGY","Score":null,"Total":0}
引用次数: 0
摘要
背景:免疫复合物介导的膜增殖性肾小球肾炎(IC-MPGN)和Alport综合征是两种不同的肾小球疾病,具有不同的病理生理机制。它们的共存极为罕见,可能会给诊断和治疗带来挑战。病例介绍:一名42岁女性,表现为持续性蛋白尿和血尿。初步实验室检查显示蛋白尿进行性增加。她有肾脏疾病的家族史。肾活检最初应患者要求推迟,随后发现弥漫性系膜和局灶性毛细血管内增生,GBM增厚,肾小球周围纤维化,IgM, C3和C1q免疫复合物沉积,从而确认IC-MPGN的诊断。基因检测发现COL4A5 c.1871G > a (p.Gly624Asp)杂合突变与x连锁Alport综合征一致。病人现在接受保守治疗。结论:本病例突出了IC-MPGN、慢性TIN和Alport综合征共存的罕见病例。进行性肾小球疾病患者,特别是有肾脏疾病家族史或肾外病变的患者,应考虑进行遗传评估。早期肾活检和基因检测对具有重叠特征的复杂病例的指导管理至关重要。
A rare case of dual glomerular pathology: Alport syndrome and immune complex-mediated MPGN.
Background: Immune complex-mediated membranoproliferative glomerulonephritis (IC-MPGN) and Alport syndrome are distinct glomerular diseases with different pathophysiologic mechanisms. Their coexistence is extremely rare and may present diagnostic and therapeutic challenges.
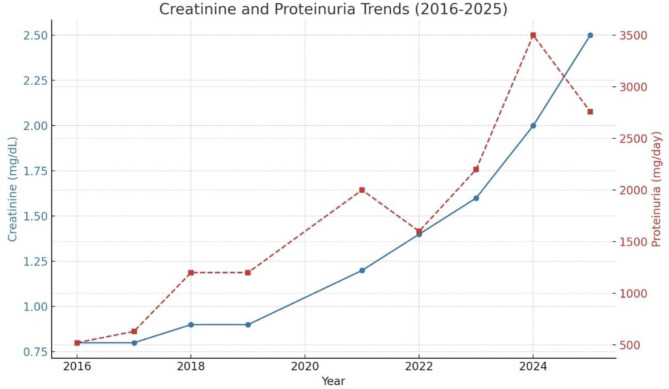
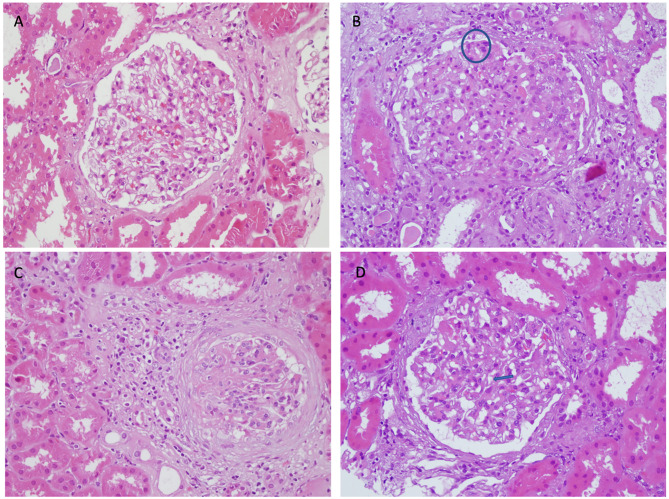
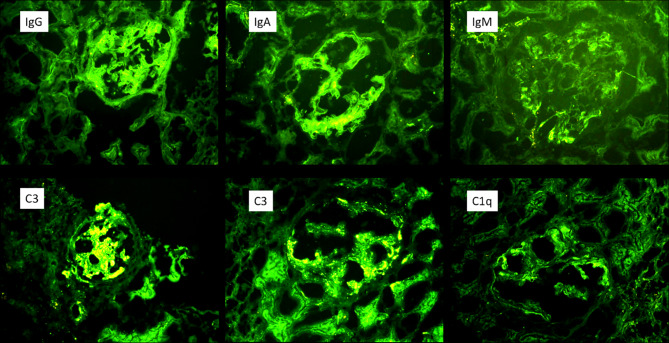
Case presentation: A 42-year-old woman presented with persistent proteinuria and hematuria. Initial laboratory evaluations showed a progressive increase in proteinuria. She had a family history of renal disease. A renal biopsy, which had initially been postponed on patient request, subsequently revealed diffuse mesangial and focal endocapillary proliferation, GBM thickening, periglomerular fibrosis, and immune complex deposition with IgM, C3, and C1q, thereby confirming the diagnosis of IC-MPGN. Genetic testing identified a heterozygous COL4A5 c.1871G > A (p.Gly624Asp) mutation consistent with X-linked Alport syndrome. The patient follows conservative management now.
Conclusion: This case highlights the rare coexistence of IC-MPGN, chronic TIN and Alport syndrome. In patients with progressive glomerular disease, especially with a family history of renal disease or extrarenal findings, genetic evaluation should be considered. Early renal biopsy and genetic testing are essential to guide management in complex cases with overlapping features.
期刊介绍:
BMC Nephrology is an open access journal publishing original peer-reviewed research articles in all aspects of the prevention, diagnosis and management of kidney and associated disorders, as well as related molecular genetics, pathophysiology, and epidemiology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


