Martyna Mocarska, Adrianna Muciek, Julia Dolinkiewicz, Anna Maria Maryńczak, Nicole Nitschke, Katarzyna Strakowska, Laura Opalska, Anna Maria Orłowska
{"title":"内瑟顿综合征:发病机制、临床表现和治疗策略的综合文献综述。","authors":"Martyna Mocarska, Adrianna Muciek, Julia Dolinkiewicz, Anna Maria Maryńczak, Nicole Nitschke, Katarzyna Strakowska, Laura Opalska, Anna Maria Orłowska","doi":"10.34763/jmotherandchild.20252901.d-25-00014","DOIUrl":null,"url":null,"abstract":"<p><p>Netherton syndrome (NS) is a rare, autosomal recessive genodermatosis resulting from mutations in the SPINK5 gene, which encodes the LEKTI (Lympho-Epithelial Kazal-type-related inhibitor) protein. This deficiency leads to dysregulated epidermal protease activity, primarily of kallikrein-related peptidases (KLKs), causing severe skin barrier defects, abnormal desquamation, and a complex immune dysregulation involving the T<sub>H</sub>2 and T<sub>H</sub>17 pathways. Clinically, NS is characterised by a triad of ichthyosiform erythroderma (often evolving from congenital ichthyosiform erythroderma to ichthyosis linearis circumflexa); pathognomonic hair shaft abnormalities, such as trichorrhexis invaginata (\"bamboo hair\"); and atopic manifestations with elevated serum IgE. Diagnosis can be challenging due to symptomatic overlap with other inflammatory dermatoses, congenital ichthyosis, and primary immunodeficiencies. Confirmation relies on clinical findings, trichoscopic hair examination, and SPINK5 genetic testing. Management is currently largely supportive, focusing on emollients, antiseptics, and cautious use of topical anti-inflammatory agents. While traditional systemic treatments have limitations, emerging targeted therapies, including biologics and gene therapy, show promise, but require further investigation through robust clinical trials to establish their efficacy and safety. This review highlights the diagnostic intricacies and evolving therapeutic landscape of this complex disorder.</p>","PeriodicalId":73842,"journal":{"name":"Journal of mother and child","volume":"29 1","pages":"106-113"},"PeriodicalIF":0.0000,"publicationDate":"2025-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12406987/pdf/","citationCount":"0","resultStr":"{\"title\":\"Netherton Syndrome: A Comprehensive Literature Review of Pathogenesis, Clinical Manifestations, and Therapeutic Strategies.\",\"authors\":\"Martyna Mocarska, Adrianna Muciek, Julia Dolinkiewicz, Anna Maria Maryńczak, Nicole Nitschke, Katarzyna Strakowska, Laura Opalska, Anna Maria Orłowska\",\"doi\":\"10.34763/jmotherandchild.20252901.d-25-00014\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Netherton syndrome (NS) is a rare, autosomal recessive genodermatosis resulting from mutations in the SPINK5 gene, which encodes the LEKTI (Lympho-Epithelial Kazal-type-related inhibitor) protein. This deficiency leads to dysregulated epidermal protease activity, primarily of kallikrein-related peptidases (KLKs), causing severe skin barrier defects, abnormal desquamation, and a complex immune dysregulation involving the T<sub>H</sub>2 and T<sub>H</sub>17 pathways. Clinically, NS is characterised by a triad of ichthyosiform erythroderma (often evolving from congenital ichthyosiform erythroderma to ichthyosis linearis circumflexa); pathognomonic hair shaft abnormalities, such as trichorrhexis invaginata (\\\"bamboo hair\\\"); and atopic manifestations with elevated serum IgE. Diagnosis can be challenging due to symptomatic overlap with other inflammatory dermatoses, congenital ichthyosis, and primary immunodeficiencies. Confirmation relies on clinical findings, trichoscopic hair examination, and SPINK5 genetic testing. Management is currently largely supportive, focusing on emollients, antiseptics, and cautious use of topical anti-inflammatory agents. While traditional systemic treatments have limitations, emerging targeted therapies, including biologics and gene therapy, show promise, but require further investigation through robust clinical trials to establish their efficacy and safety. This review highlights the diagnostic intricacies and evolving therapeutic landscape of this complex disorder.</p>\",\"PeriodicalId\":73842,\"journal\":{\"name\":\"Journal of mother and child\",\"volume\":\"29 1\",\"pages\":\"106-113\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2025-09-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12406987/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of mother and child\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.34763/jmotherandchild.20252901.d-25-00014\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/2/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of mother and child","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.34763/jmotherandchild.20252901.d-25-00014","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/2/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
Netherton Syndrome: A Comprehensive Literature Review of Pathogenesis, Clinical Manifestations, and Therapeutic Strategies.
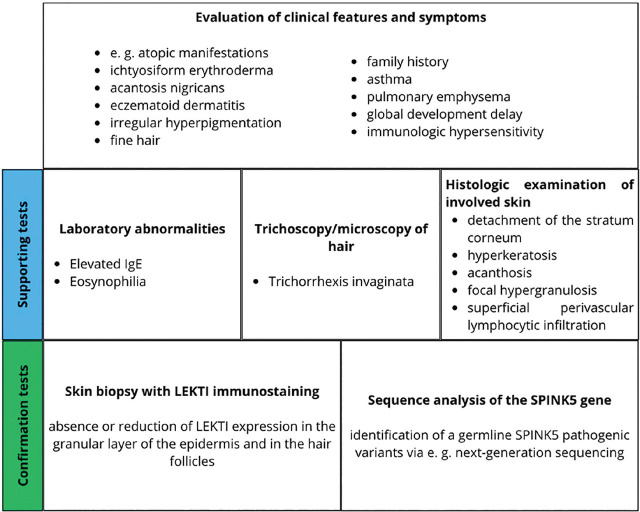
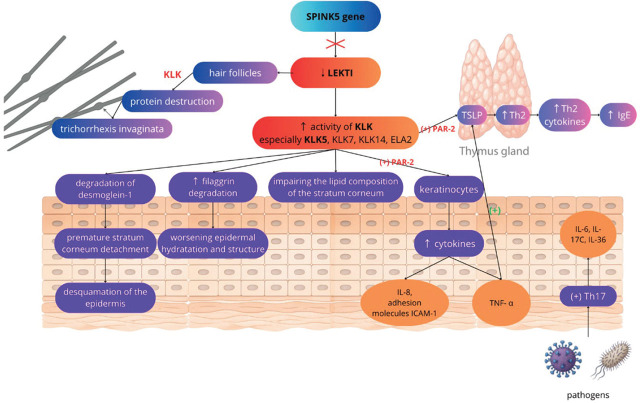
Netherton syndrome (NS) is a rare, autosomal recessive genodermatosis resulting from mutations in the SPINK5 gene, which encodes the LEKTI (Lympho-Epithelial Kazal-type-related inhibitor) protein. This deficiency leads to dysregulated epidermal protease activity, primarily of kallikrein-related peptidases (KLKs), causing severe skin barrier defects, abnormal desquamation, and a complex immune dysregulation involving the TH2 and TH17 pathways. Clinically, NS is characterised by a triad of ichthyosiform erythroderma (often evolving from congenital ichthyosiform erythroderma to ichthyosis linearis circumflexa); pathognomonic hair shaft abnormalities, such as trichorrhexis invaginata ("bamboo hair"); and atopic manifestations with elevated serum IgE. Diagnosis can be challenging due to symptomatic overlap with other inflammatory dermatoses, congenital ichthyosis, and primary immunodeficiencies. Confirmation relies on clinical findings, trichoscopic hair examination, and SPINK5 genetic testing. Management is currently largely supportive, focusing on emollients, antiseptics, and cautious use of topical anti-inflammatory agents. While traditional systemic treatments have limitations, emerging targeted therapies, including biologics and gene therapy, show promise, but require further investigation through robust clinical trials to establish their efficacy and safety. This review highlights the diagnostic intricacies and evolving therapeutic landscape of this complex disorder.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


