{"title":"2023 - 2024年宁夏地区重症急性呼吸道感染患者偏肺病毒流行病学及遗传多样性分析","authors":"Ting Mu, Jianxin Pei, Jingting Wang, Ling Niu, Zhonglan Wu","doi":"10.3390/diseases13080255","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Human metapneumovirus (HMPV) is a major pathogen responsible for causing severe acute respiratory infections (SARI). Whole-genome sequencing can better identify transmission events and outbreaks. In this study, we aimed to investigate the epidemiology and genetic diversity of HMPV in SARI cases in Ningxia, China.</p><p><strong>Methods: </strong>We collected respiratory tract samples from hospitalized patients with SARI from October 2023 to September 2024 in Ningxia, China. Nasopharyngeal swabs were tested for respiratory viruses with qRT-PCR. Whole-genome sequences were determined for samples with high viral loads using an amplicon-based method.</p><p><strong>Results: </strong>We enrolled 2873 SARI patients from October 2023 to September 2024, and found an HMPV-positive proportion of 3.06% (88/2873). Children aged 4 years were particularly susceptible to HMPV infection, with a positive proportion of 10.92% (13/119). HMPV exhibits distinct seasonal characteristics, consistent with its established epidemiological pattern, with a peak incidence occurring during winter months. Sixteen complete HMPV genome sequences were obtained. Among these, 81.25% (13/16) were identified as genotype A (A2.2.2: 92.31%, 12/13; A2.2.1: 7.69%, 1/13) and 18.75% (3/16) as genotype B1. Notably, the dominant strain was 111nt-dup in genotype A2.2.2. Sequence analysis of HMPV genes revealed divergent G-gene sequence identities between different genotypes. Additionally, the potential glycosylation sites of the G protein varied across genotypes.</p><p><strong>Conclusions: </strong>In this study, we found that the 111nt-dup strain was the dominant one in genotype A, and multiple genotypes co-circulated in Ningxia from October 2023 to September 2024. The HMPV G protein exhibited the highest level of inter-strain diversity between genotypes. These findings provide valuable insights into the prevention and control of HMPV infections in China.</p>","PeriodicalId":72832,"journal":{"name":"Diseases (Basel, Switzerland)","volume":"13 8","pages":""},"PeriodicalIF":3.0000,"publicationDate":"2025-08-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12385779/pdf/","citationCount":"0","resultStr":"{\"title\":\"Epidemiology and Genetic Diversity of Human Metapneumovirus in Patients with Severe Acute Respiratory Infection from 2023 to 2024 in Ningxia, China.\",\"authors\":\"Ting Mu, Jianxin Pei, Jingting Wang, Ling Niu, Zhonglan Wu\",\"doi\":\"10.3390/diseases13080255\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Human metapneumovirus (HMPV) is a major pathogen responsible for causing severe acute respiratory infections (SARI). Whole-genome sequencing can better identify transmission events and outbreaks. In this study, we aimed to investigate the epidemiology and genetic diversity of HMPV in SARI cases in Ningxia, China.</p><p><strong>Methods: </strong>We collected respiratory tract samples from hospitalized patients with SARI from October 2023 to September 2024 in Ningxia, China. Nasopharyngeal swabs were tested for respiratory viruses with qRT-PCR. Whole-genome sequences were determined for samples with high viral loads using an amplicon-based method.</p><p><strong>Results: </strong>We enrolled 2873 SARI patients from October 2023 to September 2024, and found an HMPV-positive proportion of 3.06% (88/2873). Children aged 4 years were particularly susceptible to HMPV infection, with a positive proportion of 10.92% (13/119). HMPV exhibits distinct seasonal characteristics, consistent with its established epidemiological pattern, with a peak incidence occurring during winter months. Sixteen complete HMPV genome sequences were obtained. Among these, 81.25% (13/16) were identified as genotype A (A2.2.2: 92.31%, 12/13; A2.2.1: 7.69%, 1/13) and 18.75% (3/16) as genotype B1. Notably, the dominant strain was 111nt-dup in genotype A2.2.2. Sequence analysis of HMPV genes revealed divergent G-gene sequence identities between different genotypes. Additionally, the potential glycosylation sites of the G protein varied across genotypes.</p><p><strong>Conclusions: </strong>In this study, we found that the 111nt-dup strain was the dominant one in genotype A, and multiple genotypes co-circulated in Ningxia from October 2023 to September 2024. The HMPV G protein exhibited the highest level of inter-strain diversity between genotypes. These findings provide valuable insights into the prevention and control of HMPV infections in China.</p>\",\"PeriodicalId\":72832,\"journal\":{\"name\":\"Diseases (Basel, Switzerland)\",\"volume\":\"13 8\",\"pages\":\"\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2025-08-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12385779/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Diseases (Basel, Switzerland)\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.3390/diseases13080255\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"MEDICINE, RESEARCH & EXPERIMENTAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Diseases (Basel, Switzerland)","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/diseases13080255","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
Epidemiology and Genetic Diversity of Human Metapneumovirus in Patients with Severe Acute Respiratory Infection from 2023 to 2024 in Ningxia, China.
Background: Human metapneumovirus (HMPV) is a major pathogen responsible for causing severe acute respiratory infections (SARI). Whole-genome sequencing can better identify transmission events and outbreaks. In this study, we aimed to investigate the epidemiology and genetic diversity of HMPV in SARI cases in Ningxia, China.
Methods: We collected respiratory tract samples from hospitalized patients with SARI from October 2023 to September 2024 in Ningxia, China. Nasopharyngeal swabs were tested for respiratory viruses with qRT-PCR. Whole-genome sequences were determined for samples with high viral loads using an amplicon-based method.
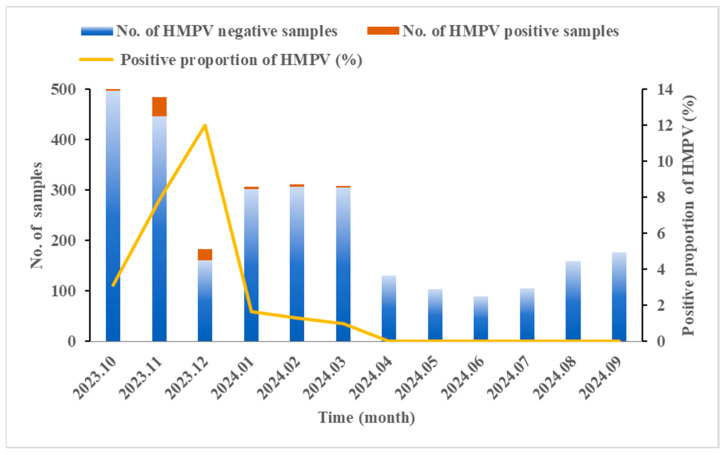
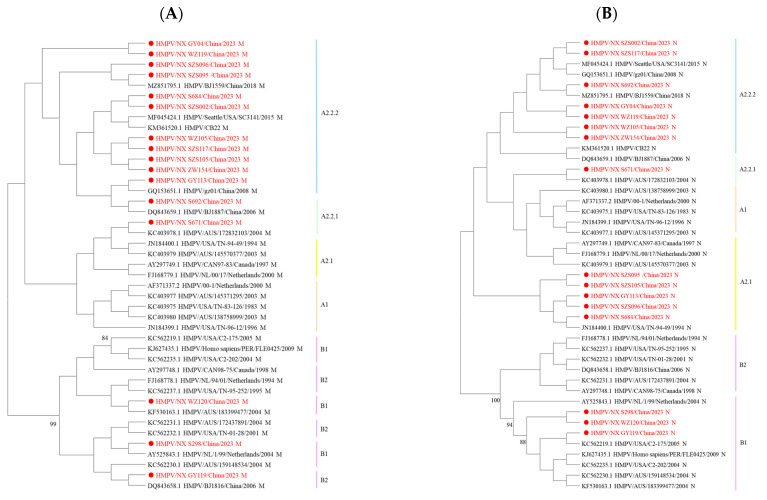
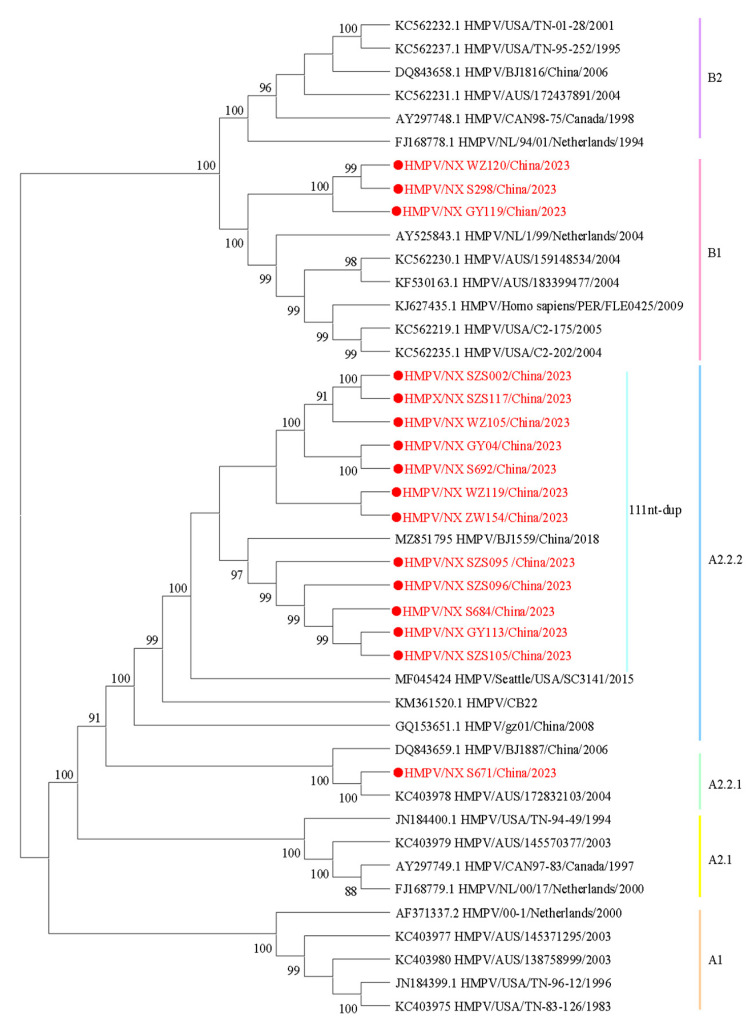
Results: We enrolled 2873 SARI patients from October 2023 to September 2024, and found an HMPV-positive proportion of 3.06% (88/2873). Children aged 4 years were particularly susceptible to HMPV infection, with a positive proportion of 10.92% (13/119). HMPV exhibits distinct seasonal characteristics, consistent with its established epidemiological pattern, with a peak incidence occurring during winter months. Sixteen complete HMPV genome sequences were obtained. Among these, 81.25% (13/16) were identified as genotype A (A2.2.2: 92.31%, 12/13; A2.2.1: 7.69%, 1/13) and 18.75% (3/16) as genotype B1. Notably, the dominant strain was 111nt-dup in genotype A2.2.2. Sequence analysis of HMPV genes revealed divergent G-gene sequence identities between different genotypes. Additionally, the potential glycosylation sites of the G protein varied across genotypes.
Conclusions: In this study, we found that the 111nt-dup strain was the dominant one in genotype A, and multiple genotypes co-circulated in Ningxia from October 2023 to September 2024. The HMPV G protein exhibited the highest level of inter-strain diversity between genotypes. These findings provide valuable insights into the prevention and control of HMPV infections in China.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


