{"title":"PMM2-CDG临床和遗传谱的研究:来自一个新变异家族和先前研究的见解。","authors":"Parnian Alagha, Tara Akhtarkhavari, Ebrahim Shokouhian, Fatemeh Ghodratpour, Sanaz Arzhangi, Hossein Najmabadi, Kimia Kahrizi","doi":"10.34172/aim.34187","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>PMM2-CDG, also known as congenital disorder of glycosylation type 1a, is the most common N-linked glycosylation disorder, characterized by a wide range of neurological and multisystem manifestations. Understanding the genotype-phenotype correlations is essential for accurate diagnosis and patient management. This study aims to identify the genetic cause of PMM2-CDG in an Iranian family with multiple affected members, and to analyze the genetic and clinical spectrum of the disorder through a comprehensive literature review.</p><p><strong>Methods: </strong>Exome sequencing re-analysis was performed to detect disease-causing variants in three affected siblings. Additionally, a literature review was conducted, analyzing 91 previously reported cases of PMM2-CDG to determine the most prevalent variants and associated clinical features.</p><p><strong>Results: </strong>A novel splice site variant (c.640-9T>A) was identified alongside a previously reported missense mutation (c.647A>T; p.N216I) in the affected individuals. The literature review revealed that the most frequent <i>PMM2</i> variants were p.R141H (28.8%), p.V231M (12.8%), p.N216I (6.4%), and p.V129M (5.8%), with 77.6% of mutations occurring in exons 5 and 8. The most common clinical findings included developmental delay, ocular abnormalities (hypertelorism, strabismus), muscular system defects (hypotonia, muscle weakness), neurological symptoms (abnormal MRI findings), cardiovascular involvement (pericarditis, pericardial effusion), and clotting disorders.</p><p><strong>Conclusion: </strong>We expect that our detailed clinical study will improve the genotype-phenotype interpretation of causal PMM2-CDG variants and the analysis of next-generation sequencing data, leading to clarification of the cause of complicated cases of rare diseases.</p>","PeriodicalId":55469,"journal":{"name":"Archives of Iranian Medicine","volume":"28 7","pages":"387-397"},"PeriodicalIF":1.0000,"publicationDate":"2025-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12435617/pdf/","citationCount":"0","resultStr":"{\"title\":\"Investigation of the Clinical and Genetic Spectrum of PMM2-CDG: Insights from a Family with a Novel Variant and Previous Studies.\",\"authors\":\"Parnian Alagha, Tara Akhtarkhavari, Ebrahim Shokouhian, Fatemeh Ghodratpour, Sanaz Arzhangi, Hossein Najmabadi, Kimia Kahrizi\",\"doi\":\"10.34172/aim.34187\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>PMM2-CDG, also known as congenital disorder of glycosylation type 1a, is the most common N-linked glycosylation disorder, characterized by a wide range of neurological and multisystem manifestations. Understanding the genotype-phenotype correlations is essential for accurate diagnosis and patient management. This study aims to identify the genetic cause of PMM2-CDG in an Iranian family with multiple affected members, and to analyze the genetic and clinical spectrum of the disorder through a comprehensive literature review.</p><p><strong>Methods: </strong>Exome sequencing re-analysis was performed to detect disease-causing variants in three affected siblings. Additionally, a literature review was conducted, analyzing 91 previously reported cases of PMM2-CDG to determine the most prevalent variants and associated clinical features.</p><p><strong>Results: </strong>A novel splice site variant (c.640-9T>A) was identified alongside a previously reported missense mutation (c.647A>T; p.N216I) in the affected individuals. The literature review revealed that the most frequent <i>PMM2</i> variants were p.R141H (28.8%), p.V231M (12.8%), p.N216I (6.4%), and p.V129M (5.8%), with 77.6% of mutations occurring in exons 5 and 8. The most common clinical findings included developmental delay, ocular abnormalities (hypertelorism, strabismus), muscular system defects (hypotonia, muscle weakness), neurological symptoms (abnormal MRI findings), cardiovascular involvement (pericarditis, pericardial effusion), and clotting disorders.</p><p><strong>Conclusion: </strong>We expect that our detailed clinical study will improve the genotype-phenotype interpretation of causal PMM2-CDG variants and the analysis of next-generation sequencing data, leading to clarification of the cause of complicated cases of rare diseases.</p>\",\"PeriodicalId\":55469,\"journal\":{\"name\":\"Archives of Iranian Medicine\",\"volume\":\"28 7\",\"pages\":\"387-397\"},\"PeriodicalIF\":1.0000,\"publicationDate\":\"2025-07-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12435617/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Archives of Iranian Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.34172/aim.34187\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"MEDICINE, GENERAL & INTERNAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Archives of Iranian Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.34172/aim.34187","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"MEDICINE, GENERAL & INTERNAL","Score":null,"Total":0}
Investigation of the Clinical and Genetic Spectrum of PMM2-CDG: Insights from a Family with a Novel Variant and Previous Studies.
Background: PMM2-CDG, also known as congenital disorder of glycosylation type 1a, is the most common N-linked glycosylation disorder, characterized by a wide range of neurological and multisystem manifestations. Understanding the genotype-phenotype correlations is essential for accurate diagnosis and patient management. This study aims to identify the genetic cause of PMM2-CDG in an Iranian family with multiple affected members, and to analyze the genetic and clinical spectrum of the disorder through a comprehensive literature review.
Methods: Exome sequencing re-analysis was performed to detect disease-causing variants in three affected siblings. Additionally, a literature review was conducted, analyzing 91 previously reported cases of PMM2-CDG to determine the most prevalent variants and associated clinical features.
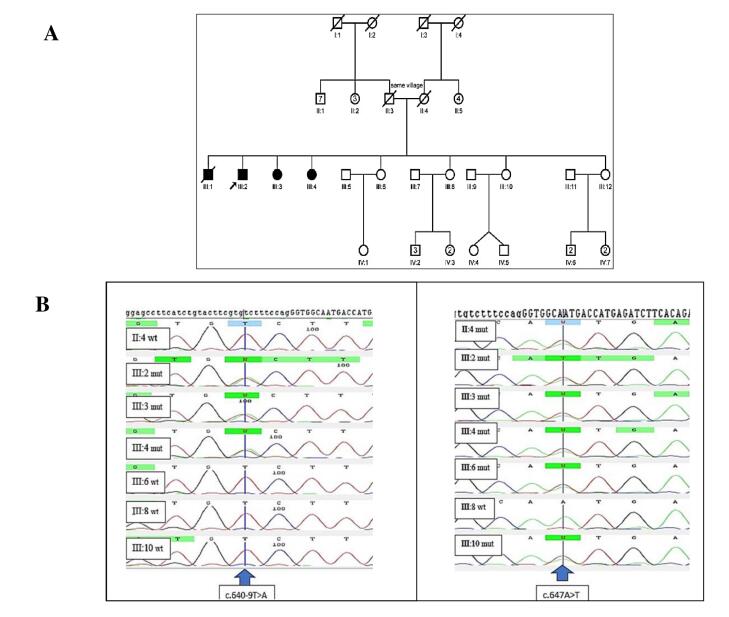
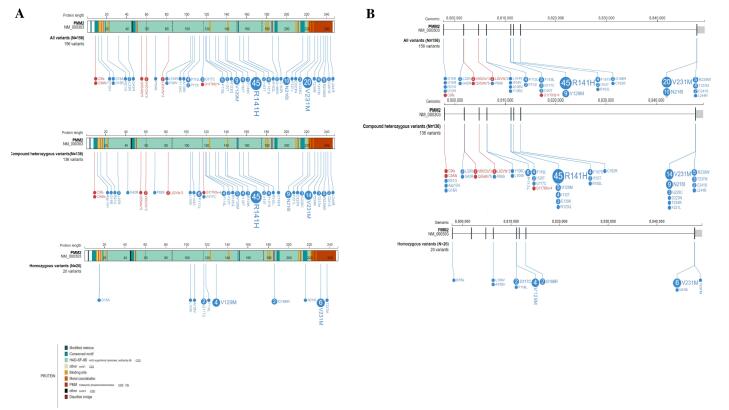
Results: A novel splice site variant (c.640-9T>A) was identified alongside a previously reported missense mutation (c.647A>T; p.N216I) in the affected individuals. The literature review revealed that the most frequent PMM2 variants were p.R141H (28.8%), p.V231M (12.8%), p.N216I (6.4%), and p.V129M (5.8%), with 77.6% of mutations occurring in exons 5 and 8. The most common clinical findings included developmental delay, ocular abnormalities (hypertelorism, strabismus), muscular system defects (hypotonia, muscle weakness), neurological symptoms (abnormal MRI findings), cardiovascular involvement (pericarditis, pericardial effusion), and clotting disorders.
Conclusion: We expect that our detailed clinical study will improve the genotype-phenotype interpretation of causal PMM2-CDG variants and the analysis of next-generation sequencing data, leading to clarification of the cause of complicated cases of rare diseases.
期刊介绍:
Aim and Scope: The Archives of Iranian Medicine (AIM) is a monthly peer-reviewed multidisciplinary medical publication. The journal welcomes contributions particularly relevant to the Middle-East region and publishes biomedical experiences and clinical investigations on prevalent diseases in the region as well as analyses of factors that may modulate the incidence, course, and management of diseases and pertinent medical problems. Manuscripts with didactic orientation and subjects exclusively of local interest will not be considered for publication.The 2016 Impact Factor of "Archives of Iranian Medicine" is 1.20.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


