Niel Verbrigghe, Hilde Muylle, Marie Pegard, Hendrik Rietman, Vuk Đorđević, Marina Ćeran, Isabel Roldán-Ruiz
{"title":"在基因组预测和机器学习- gwas工作流程中分离大豆GxE效应。","authors":"Niel Verbrigghe, Hilde Muylle, Marie Pegard, Hendrik Rietman, Vuk Đorđević, Marina Ćeran, Isabel Roldán-Ruiz","doi":"10.1186/s13007-025-01434-0","DOIUrl":null,"url":null,"abstract":"<p><p>Integrating genotype-by-Environment (GxE) interactions into genomic prediction models has been demonstrated to enhance the accuracy of predictions for crops exposed to unfavourable environmental conditions. However, despite the increasing complexity of machine learning models in genomic prediction, no model or approach has been found to be overall superior in comparison to a classical genomic best linear unbiased prediction (GBLUP) model. In this paper, we compared two GBLUP models (Linear Mixed Effects model and Bayesian GBLUP) with two machine learning models (Random Forest and Extreme Gradient Boosting) on the EUCLEG soybean genotype set phenotyped in Belgium and Serbia. We found similar performance for the Bayesian GBLUP and the two machine learning methods. However, using a workflow that decomposed the environment-specific BLUPs into a main genetic and an interaction GxE effect, we found increased predictive ability for the interaction component compared to a single-component approach. Furthermore, conducting a machine learning-genome wide association study (ML-GWAS) on both components allowed us to identify important markers for the main genetic effect, as well as environment-specific markers. These could then be associated with correlated markers in other environments. By constructing a small random forest model using only 50 uncorrelated, important markers we constructed a genomic prediction model with similar predictive ability over all scenarios when compared to the large models including all markers. The results demonstrate a new, integrated genomic prediction and machine learning-genome-wide association study (ML-GWAS) approach, aimed at high predictive ability and coupled marker detection in the soybean genome for traits phenotyped in different environments.</p>","PeriodicalId":20100,"journal":{"name":"Plant Methods","volume":"21 1","pages":"119"},"PeriodicalIF":4.4000,"publicationDate":"2025-08-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12376716/pdf/","citationCount":"0","resultStr":"{\"title\":\"Disentangling soybean GxE effects in an integrated genomic prediction and machine learning-GWAS workflow.\",\"authors\":\"Niel Verbrigghe, Hilde Muylle, Marie Pegard, Hendrik Rietman, Vuk Đorđević, Marina Ćeran, Isabel Roldán-Ruiz\",\"doi\":\"10.1186/s13007-025-01434-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Integrating genotype-by-Environment (GxE) interactions into genomic prediction models has been demonstrated to enhance the accuracy of predictions for crops exposed to unfavourable environmental conditions. However, despite the increasing complexity of machine learning models in genomic prediction, no model or approach has been found to be overall superior in comparison to a classical genomic best linear unbiased prediction (GBLUP) model. In this paper, we compared two GBLUP models (Linear Mixed Effects model and Bayesian GBLUP) with two machine learning models (Random Forest and Extreme Gradient Boosting) on the EUCLEG soybean genotype set phenotyped in Belgium and Serbia. We found similar performance for the Bayesian GBLUP and the two machine learning methods. However, using a workflow that decomposed the environment-specific BLUPs into a main genetic and an interaction GxE effect, we found increased predictive ability for the interaction component compared to a single-component approach. Furthermore, conducting a machine learning-genome wide association study (ML-GWAS) on both components allowed us to identify important markers for the main genetic effect, as well as environment-specific markers. These could then be associated with correlated markers in other environments. By constructing a small random forest model using only 50 uncorrelated, important markers we constructed a genomic prediction model with similar predictive ability over all scenarios when compared to the large models including all markers. The results demonstrate a new, integrated genomic prediction and machine learning-genome-wide association study (ML-GWAS) approach, aimed at high predictive ability and coupled marker detection in the soybean genome for traits phenotyped in different environments.</p>\",\"PeriodicalId\":20100,\"journal\":{\"name\":\"Plant Methods\",\"volume\":\"21 1\",\"pages\":\"119\"},\"PeriodicalIF\":4.4000,\"publicationDate\":\"2025-08-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12376716/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Plant Methods\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s13007-025-01434-0\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Plant Methods","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13007-025-01434-0","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Disentangling soybean GxE effects in an integrated genomic prediction and machine learning-GWAS workflow.
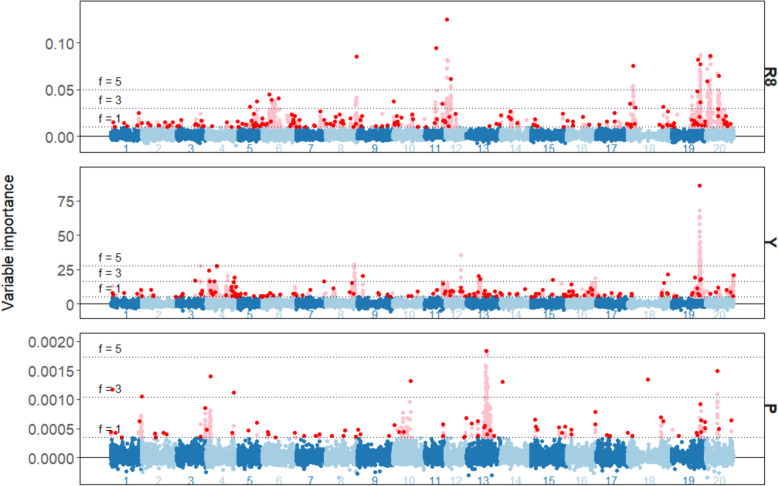
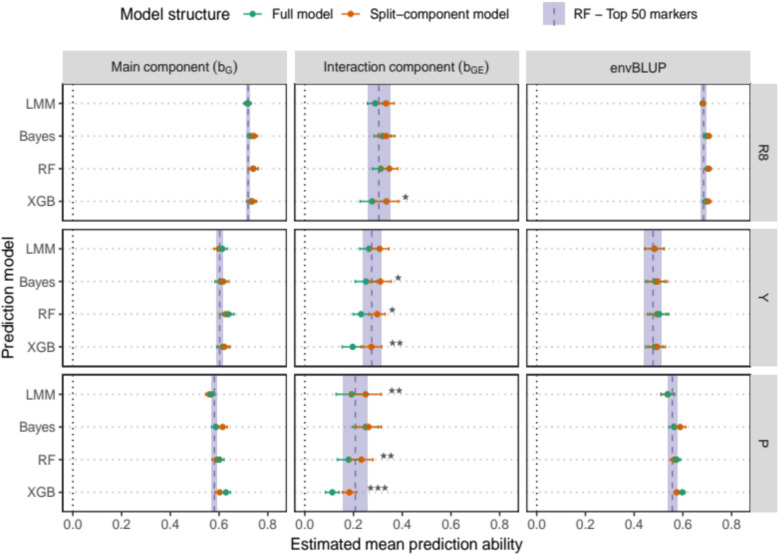
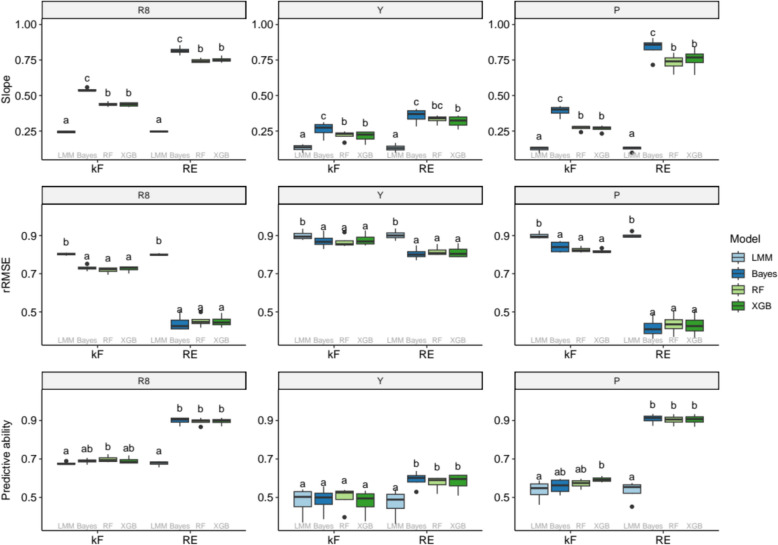
Integrating genotype-by-Environment (GxE) interactions into genomic prediction models has been demonstrated to enhance the accuracy of predictions for crops exposed to unfavourable environmental conditions. However, despite the increasing complexity of machine learning models in genomic prediction, no model or approach has been found to be overall superior in comparison to a classical genomic best linear unbiased prediction (GBLUP) model. In this paper, we compared two GBLUP models (Linear Mixed Effects model and Bayesian GBLUP) with two machine learning models (Random Forest and Extreme Gradient Boosting) on the EUCLEG soybean genotype set phenotyped in Belgium and Serbia. We found similar performance for the Bayesian GBLUP and the two machine learning methods. However, using a workflow that decomposed the environment-specific BLUPs into a main genetic and an interaction GxE effect, we found increased predictive ability for the interaction component compared to a single-component approach. Furthermore, conducting a machine learning-genome wide association study (ML-GWAS) on both components allowed us to identify important markers for the main genetic effect, as well as environment-specific markers. These could then be associated with correlated markers in other environments. By constructing a small random forest model using only 50 uncorrelated, important markers we constructed a genomic prediction model with similar predictive ability over all scenarios when compared to the large models including all markers. The results demonstrate a new, integrated genomic prediction and machine learning-genome-wide association study (ML-GWAS) approach, aimed at high predictive ability and coupled marker detection in the soybean genome for traits phenotyped in different environments.
期刊介绍:
Plant Methods is an open access, peer-reviewed, online journal for the plant research community that encompasses all aspects of technological innovation in the plant sciences.
There is no doubt that we have entered an exciting new era in plant biology. The completion of the Arabidopsis genome sequence, and the rapid progress being made in other plant genomics projects are providing unparalleled opportunities for progress in all areas of plant science. Nevertheless, enormous challenges lie ahead if we are to understand the function of every gene in the genome, and how the individual parts work together to make the whole organism. Achieving these goals will require an unprecedented collaborative effort, combining high-throughput, system-wide technologies with more focused approaches that integrate traditional disciplines such as cell biology, biochemistry and molecular genetics.
Technological innovation is probably the most important catalyst for progress in any scientific discipline. Plant Methods’ goal is to stimulate the development and adoption of new and improved techniques and research tools and, where appropriate, to promote consistency of methodologies for better integration of data from different laboratories.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


