Ekram Fateen, Soha S Nosier, Nahla N Abdel Aziz, Amira M Radwan, Eman E A Mohammed
{"title":"临床,生化和分子特征的三filippo综合征(MPS IIIA)在一组埃及患者。","authors":"Ekram Fateen, Soha S Nosier, Nahla N Abdel Aziz, Amira M Radwan, Eman E A Mohammed","doi":"10.1186/s13023-025-03971-2","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Lysosomal storage diseases (LSDs) is a large group of genetically heterogeneous inherited metabolic disorders that affect the functions of the lysosomes in various human tissues. Mucopolysaccharidosis type IIIA (MPSIIIA), Sanflippo syndrome A, is a rare autosomal recessive LSD caused by biallelic variants in the SGSH gene, codes for the lysosomal enzyme heparan-N-sulphatase. This study aimed to find out the SGSH mutational spectrum, clinical and biochemical characteristics in a cohort of MPS IIIA Egyptian patients.</p><p><strong>Results: </strong>Ten patients derived from 9 unrelated families, clinically and biochemically diagnosed having MPS IIIA secondary to heparan sulphatase deficiency, were enrolled. Patients, variably, displayed early-onset and progressive neurological and mental deterioration, aggressive and hyperactive behaviors, sleep disturbances and visceromegaly. Sanger sequencing of the SGSH coding and exon-intron boundaries revealed four homozygous disease-causing variants in all the patients (100%), three previously reported (p.Y224*, p.R377C, and p.V361Sfs*52), and a novel one (c.948delA; p.D317Tfs*96). The p.Y224* in exon 6 was the most recurrent variant (5/10, 50%), followed by the missense R377C in exon 8 (3/10; 30%), while the two frameshift truncating variants, each appeared in only one patient; presenting 10% of the disease causing variants.</p><p><strong>Conclusions: </strong>The pattern of variants recurrence in unrelated Egyptian patients highlights exons 6 and 8 as hot spots for first variant screening. The molecular findings of this study expand the SGSH variant spectrum and underline specific exons for first screening of MPS IIIA patients, which would largely help the early diagnosis and genetic counselling. To the best of our knowledge, the present study is the first delineating the SGSH variant profile in Egyptian Sanflippo A patients.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"454"},"PeriodicalIF":3.5000,"publicationDate":"2025-08-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12379534/pdf/","citationCount":"0","resultStr":"{\"title\":\"Clinical, biochemical, and molecular characteristics of Sanfilippo a syndrome (MPS IIIA) in a cohort of Egyptian patients.\",\"authors\":\"Ekram Fateen, Soha S Nosier, Nahla N Abdel Aziz, Amira M Radwan, Eman E A Mohammed\",\"doi\":\"10.1186/s13023-025-03971-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Lysosomal storage diseases (LSDs) is a large group of genetically heterogeneous inherited metabolic disorders that affect the functions of the lysosomes in various human tissues. Mucopolysaccharidosis type IIIA (MPSIIIA), Sanflippo syndrome A, is a rare autosomal recessive LSD caused by biallelic variants in the SGSH gene, codes for the lysosomal enzyme heparan-N-sulphatase. This study aimed to find out the SGSH mutational spectrum, clinical and biochemical characteristics in a cohort of MPS IIIA Egyptian patients.</p><p><strong>Results: </strong>Ten patients derived from 9 unrelated families, clinically and biochemically diagnosed having MPS IIIA secondary to heparan sulphatase deficiency, were enrolled. Patients, variably, displayed early-onset and progressive neurological and mental deterioration, aggressive and hyperactive behaviors, sleep disturbances and visceromegaly. Sanger sequencing of the SGSH coding and exon-intron boundaries revealed four homozygous disease-causing variants in all the patients (100%), three previously reported (p.Y224*, p.R377C, and p.V361Sfs*52), and a novel one (c.948delA; p.D317Tfs*96). The p.Y224* in exon 6 was the most recurrent variant (5/10, 50%), followed by the missense R377C in exon 8 (3/10; 30%), while the two frameshift truncating variants, each appeared in only one patient; presenting 10% of the disease causing variants.</p><p><strong>Conclusions: </strong>The pattern of variants recurrence in unrelated Egyptian patients highlights exons 6 and 8 as hot spots for first variant screening. The molecular findings of this study expand the SGSH variant spectrum and underline specific exons for first screening of MPS IIIA patients, which would largely help the early diagnosis and genetic counselling. To the best of our knowledge, the present study is the first delineating the SGSH variant profile in Egyptian Sanflippo A patients.</p>\",\"PeriodicalId\":19651,\"journal\":{\"name\":\"Orphanet Journal of Rare Diseases\",\"volume\":\"20 1\",\"pages\":\"454\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2025-08-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12379534/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Orphanet Journal of Rare Diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13023-025-03971-2\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-03971-2","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Clinical, biochemical, and molecular characteristics of Sanfilippo a syndrome (MPS IIIA) in a cohort of Egyptian patients.
Background: Lysosomal storage diseases (LSDs) is a large group of genetically heterogeneous inherited metabolic disorders that affect the functions of the lysosomes in various human tissues. Mucopolysaccharidosis type IIIA (MPSIIIA), Sanflippo syndrome A, is a rare autosomal recessive LSD caused by biallelic variants in the SGSH gene, codes for the lysosomal enzyme heparan-N-sulphatase. This study aimed to find out the SGSH mutational spectrum, clinical and biochemical characteristics in a cohort of MPS IIIA Egyptian patients.
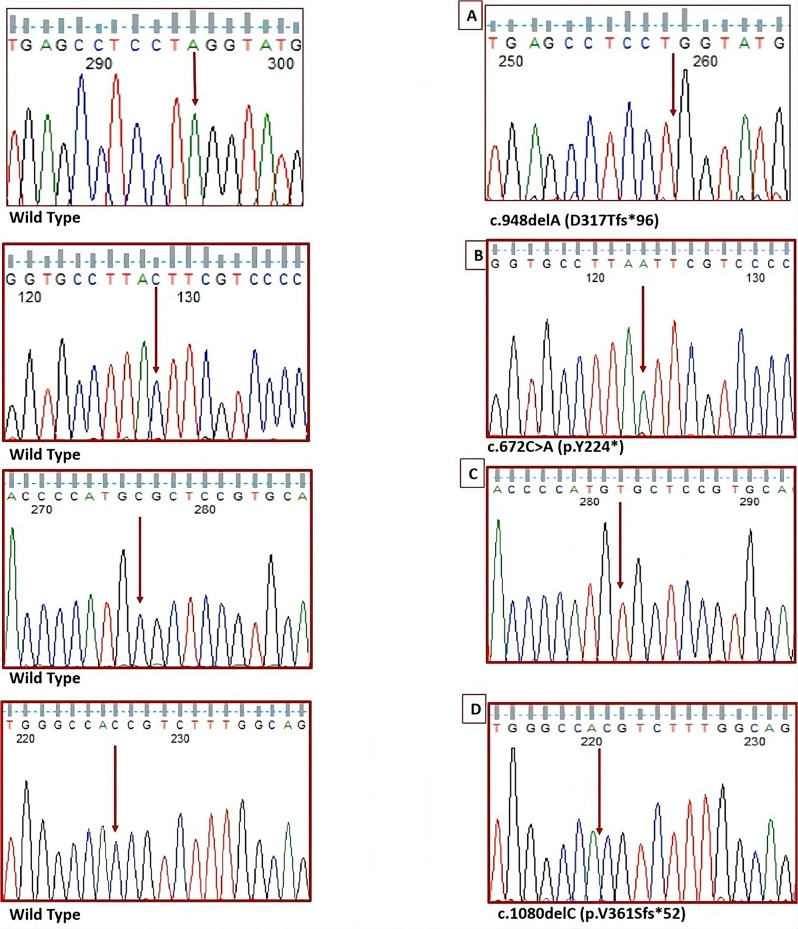
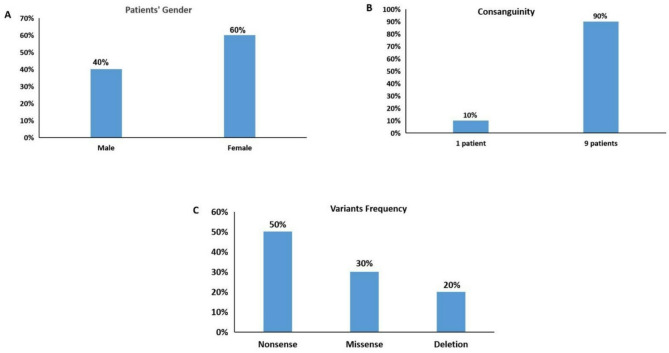
Results: Ten patients derived from 9 unrelated families, clinically and biochemically diagnosed having MPS IIIA secondary to heparan sulphatase deficiency, were enrolled. Patients, variably, displayed early-onset and progressive neurological and mental deterioration, aggressive and hyperactive behaviors, sleep disturbances and visceromegaly. Sanger sequencing of the SGSH coding and exon-intron boundaries revealed four homozygous disease-causing variants in all the patients (100%), three previously reported (p.Y224*, p.R377C, and p.V361Sfs*52), and a novel one (c.948delA; p.D317Tfs*96). The p.Y224* in exon 6 was the most recurrent variant (5/10, 50%), followed by the missense R377C in exon 8 (3/10; 30%), while the two frameshift truncating variants, each appeared in only one patient; presenting 10% of the disease causing variants.
Conclusions: The pattern of variants recurrence in unrelated Egyptian patients highlights exons 6 and 8 as hot spots for first variant screening. The molecular findings of this study expand the SGSH variant spectrum and underline specific exons for first screening of MPS IIIA patients, which would largely help the early diagnosis and genetic counselling. To the best of our knowledge, the present study is the first delineating the SGSH variant profile in Egyptian Sanflippo A patients.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


