Karli Shelton, Phu Dang, Courtney McCorkle, Pooja Mallipaddi, Nicholas Hollman, Jeremy Pomeroy, Jesse Richards
{"title":"Bardet-Biedl综合征的先天性黑素细胞痣。","authors":"Karli Shelton, Phu Dang, Courtney McCorkle, Pooja Mallipaddi, Nicholas Hollman, Jeremy Pomeroy, Jesse Richards","doi":"10.1186/s13023-025-03870-6","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Bardet-Biedl Syndrome (BBS) is a rare obesogenic disorder affecting multiple organs. The diagnosis of BBS is usually difficult and delayed due to this syndrome's wide variety of clinical features. This study aims to assess the rate of congenital melanocytic nevi (CMN) in the BBS population in an effort to bring light to an easily assessable and early manifestation of BBS to aid in earlier diagnosis.</p><p><strong>Methods: </strong>We utilized a survey distributed to patients with BBS registered within the Clinical Registry Investigating Bardet-Biedl Syndrome database. Analysis was performed to identify participants with CMN and their prevalence of major and minor symptoms of the diagnostic criteria for BBS.</p><p><strong>Results: </strong>Data from 67 patients with BBS were gathered from our surveys. Of those participants, 23.9% reported having a CMN. Patients with CMN were more likely to have abnormal reproductive health issues, high arched palate, missing teeth, dental crowning, short teeth roots, and webbed fingers and toes.</p><p><strong>Conclusion: </strong>Our findings suggest that BBS is associated with CMN, possibly through altered neural crest cell migration. Screening for CMN shows a promise as a potential non-invasive screening tool to aid in earlier diagnosis of BBS.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"462"},"PeriodicalIF":3.5000,"publicationDate":"2025-08-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12395923/pdf/","citationCount":"0","resultStr":"{\"title\":\"Congenital melanocytic nevi in Bardet-Biedl syndrome.\",\"authors\":\"Karli Shelton, Phu Dang, Courtney McCorkle, Pooja Mallipaddi, Nicholas Hollman, Jeremy Pomeroy, Jesse Richards\",\"doi\":\"10.1186/s13023-025-03870-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Bardet-Biedl Syndrome (BBS) is a rare obesogenic disorder affecting multiple organs. The diagnosis of BBS is usually difficult and delayed due to this syndrome's wide variety of clinical features. This study aims to assess the rate of congenital melanocytic nevi (CMN) in the BBS population in an effort to bring light to an easily assessable and early manifestation of BBS to aid in earlier diagnosis.</p><p><strong>Methods: </strong>We utilized a survey distributed to patients with BBS registered within the Clinical Registry Investigating Bardet-Biedl Syndrome database. Analysis was performed to identify participants with CMN and their prevalence of major and minor symptoms of the diagnostic criteria for BBS.</p><p><strong>Results: </strong>Data from 67 patients with BBS were gathered from our surveys. Of those participants, 23.9% reported having a CMN. Patients with CMN were more likely to have abnormal reproductive health issues, high arched palate, missing teeth, dental crowning, short teeth roots, and webbed fingers and toes.</p><p><strong>Conclusion: </strong>Our findings suggest that BBS is associated with CMN, possibly through altered neural crest cell migration. Screening for CMN shows a promise as a potential non-invasive screening tool to aid in earlier diagnosis of BBS.</p>\",\"PeriodicalId\":19651,\"journal\":{\"name\":\"Orphanet Journal of Rare Diseases\",\"volume\":\"20 1\",\"pages\":\"462\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2025-08-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12395923/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Orphanet Journal of Rare Diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13023-025-03870-6\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-03870-6","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Congenital melanocytic nevi in Bardet-Biedl syndrome.
Background: Bardet-Biedl Syndrome (BBS) is a rare obesogenic disorder affecting multiple organs. The diagnosis of BBS is usually difficult and delayed due to this syndrome's wide variety of clinical features. This study aims to assess the rate of congenital melanocytic nevi (CMN) in the BBS population in an effort to bring light to an easily assessable and early manifestation of BBS to aid in earlier diagnosis.
Methods: We utilized a survey distributed to patients with BBS registered within the Clinical Registry Investigating Bardet-Biedl Syndrome database. Analysis was performed to identify participants with CMN and their prevalence of major and minor symptoms of the diagnostic criteria for BBS.
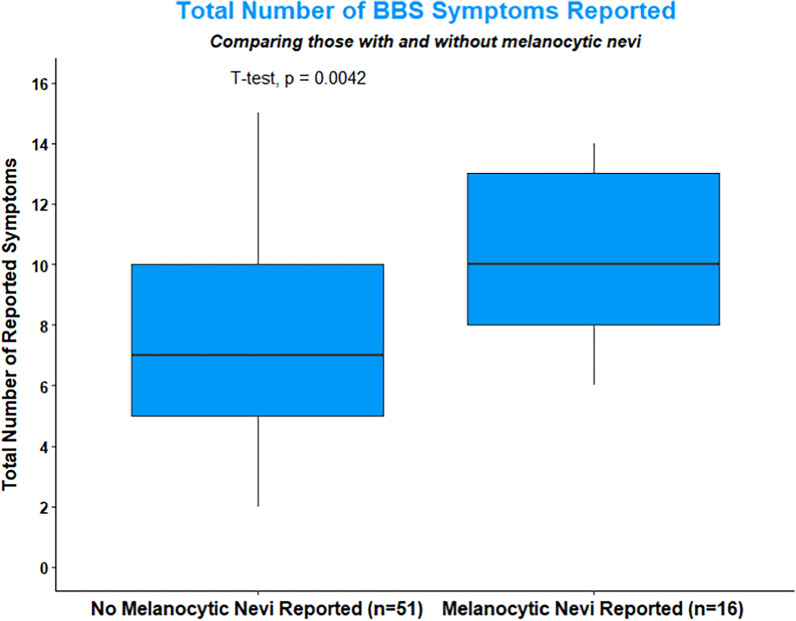
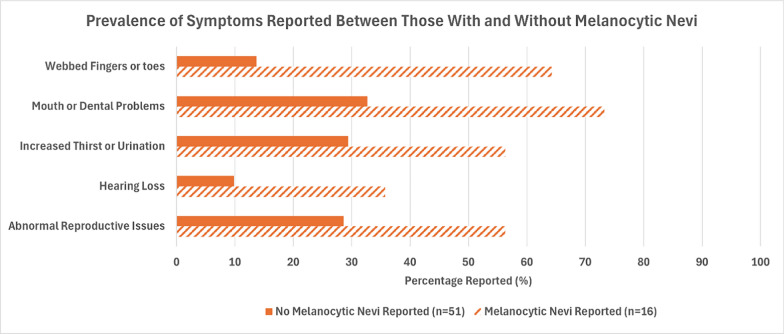
Results: Data from 67 patients with BBS were gathered from our surveys. Of those participants, 23.9% reported having a CMN. Patients with CMN were more likely to have abnormal reproductive health issues, high arched palate, missing teeth, dental crowning, short teeth roots, and webbed fingers and toes.
Conclusion: Our findings suggest that BBS is associated with CMN, possibly through altered neural crest cell migration. Screening for CMN shows a promise as a potential non-invasive screening tool to aid in earlier diagnosis of BBS.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


