Giuseppe d'Orsi, Maria Teresa Di Claudio, Antonella Liantonio, Paola Imbrici, Cosimo Damiano Altomare, Orazio Palumbo, Pietro Palumbo, Mario Benvenuto, Nicola Gambacorta, Graziano Lolli, Massimo Carella
{"title":"拉福拉病的临床过程和管理挑战:阿普利亚队列的叙事分析。","authors":"Giuseppe d'Orsi, Maria Teresa Di Claudio, Antonella Liantonio, Paola Imbrici, Cosimo Damiano Altomare, Orazio Palumbo, Pietro Palumbo, Mario Benvenuto, Nicola Gambacorta, Graziano Lolli, Massimo Carella","doi":"10.1186/s13023-025-03976-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Lafora disease (LD) is an ultra-rare, autosomal recessive neurodegenerative disorder characterized by the accumulation of Lafora bodies in the brain, leading to drug-resistant epilepsy, myoclonus, progressive dementia, and cerebellar dysfunction. This retrospective study describes the clinical course and management challenges of LD in a cohort of patients from the Apulia region of Southern Italy, where the disease prevalence appears to be higher than in other populations.</p><p><strong>Methods: </strong>We retrospectively analyzed clinical, electroencephalographic, and management data from six unrelated families with a confirmed diagnosis of LD, followed at the Neurology Unit of the Scientific Institute Casa Sollievo della Sofferenza Hospital between 2010 and 2024. Demographic information, clinical presentation, treatment history, disease progression, and outcomes were collected.</p><p><strong>Results: </strong>Our analysis identified three distinct electroclinical stages: an initial Presenting Symptoms Stage with the onset of seizures and subsequent development of myoclonus; a Progressive Neurodegeneration Stage characterized by drug-resistant epilepsy, dementia, and ataxia; and a Terminal Stage marked by severe disability, frequent seizure emergencies, and medical complications. Management in the late stages proved particularly challenging, requiring a multidisciplinary approach to address refractory seizures, status epilepticus, and medical complications such as aspiration pneumonia and respiratory failure. Home-based care, with specialized team support, played a crucial role in minimizing hospitalizations.</p><p><strong>Discussion: </strong>Our findings underscore the importance of early diagnosis and a multidisciplinary approach in the management of LD. The late stages of the disease are characterized by significant clinical challenges necessitating close collaboration among neurologists, epileptologists, and other healthcare professionals, supported by effective home-based care. The apparent higher prevalence in Apulia warrants further investigation into potential genetic or environmental factors.</p><p><strong>Conclusion: </strong>This study highlights the significant clinical burden of LD and emphasizes the importance of multidisciplinary management, particularly in the advanced stages. Home-based care supported by specialized teams and caregivers is essential for optimizing patient well-being. Further research is needed to identify early biomarkers and develop targeted therapies for this devastating condition.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"447"},"PeriodicalIF":3.5000,"publicationDate":"2025-08-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12369230/pdf/","citationCount":"0","resultStr":"{\"title\":\"Clinical course and management challenges in Lafora disease: a narrative analysis in an Apulian cohort.\",\"authors\":\"Giuseppe d'Orsi, Maria Teresa Di Claudio, Antonella Liantonio, Paola Imbrici, Cosimo Damiano Altomare, Orazio Palumbo, Pietro Palumbo, Mario Benvenuto, Nicola Gambacorta, Graziano Lolli, Massimo Carella\",\"doi\":\"10.1186/s13023-025-03976-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Lafora disease (LD) is an ultra-rare, autosomal recessive neurodegenerative disorder characterized by the accumulation of Lafora bodies in the brain, leading to drug-resistant epilepsy, myoclonus, progressive dementia, and cerebellar dysfunction. This retrospective study describes the clinical course and management challenges of LD in a cohort of patients from the Apulia region of Southern Italy, where the disease prevalence appears to be higher than in other populations.</p><p><strong>Methods: </strong>We retrospectively analyzed clinical, electroencephalographic, and management data from six unrelated families with a confirmed diagnosis of LD, followed at the Neurology Unit of the Scientific Institute Casa Sollievo della Sofferenza Hospital between 2010 and 2024. Demographic information, clinical presentation, treatment history, disease progression, and outcomes were collected.</p><p><strong>Results: </strong>Our analysis identified three distinct electroclinical stages: an initial Presenting Symptoms Stage with the onset of seizures and subsequent development of myoclonus; a Progressive Neurodegeneration Stage characterized by drug-resistant epilepsy, dementia, and ataxia; and a Terminal Stage marked by severe disability, frequent seizure emergencies, and medical complications. Management in the late stages proved particularly challenging, requiring a multidisciplinary approach to address refractory seizures, status epilepticus, and medical complications such as aspiration pneumonia and respiratory failure. Home-based care, with specialized team support, played a crucial role in minimizing hospitalizations.</p><p><strong>Discussion: </strong>Our findings underscore the importance of early diagnosis and a multidisciplinary approach in the management of LD. The late stages of the disease are characterized by significant clinical challenges necessitating close collaboration among neurologists, epileptologists, and other healthcare professionals, supported by effective home-based care. The apparent higher prevalence in Apulia warrants further investigation into potential genetic or environmental factors.</p><p><strong>Conclusion: </strong>This study highlights the significant clinical burden of LD and emphasizes the importance of multidisciplinary management, particularly in the advanced stages. Home-based care supported by specialized teams and caregivers is essential for optimizing patient well-being. Further research is needed to identify early biomarkers and develop targeted therapies for this devastating condition.</p>\",\"PeriodicalId\":19651,\"journal\":{\"name\":\"Orphanet Journal of Rare Diseases\",\"volume\":\"20 1\",\"pages\":\"447\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2025-08-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12369230/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Orphanet Journal of Rare Diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13023-025-03976-x\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-03976-x","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
背景:拉福拉病(Lafora disease, LD)是一种超罕见的常染色体隐性神经退行性疾病,其特征是拉福拉体在大脑中积聚,导致耐药癫痫、肌阵挛、进行性痴呆和小脑功能障碍。本回顾性研究描述了一组来自意大利南部普利亚地区的LD患者的临床病程和管理挑战,该地区的患病率似乎高于其他人群。方法:我们回顾性分析了2010年至2024年在Casa Sollievo della Sofferenza科学研究所神经内科就诊的6个确诊为LD的无血缘关系家庭的临床、脑电图和管理数据。收集了人口统计信息、临床表现、治疗史、疾病进展和结果。结果:我们的分析确定了三个不同的电临床阶段:最初的表现症状阶段,癫痫发作,随后发展为肌阵挛;以耐药癫痫、痴呆和共济失调为特征的进行性神经变性期;以及以严重残疾、频繁突发癫痫和医疗并发症为特征的晚期。晚期的治疗尤其具有挑战性,需要多学科的方法来解决难治性癫痫发作、癫痫持续状态以及吸入性肺炎和呼吸衰竭等医疗并发症。在专业团队的支持下,家庭护理在尽量减少住院方面发挥了关键作用。讨论:我们的研究结果强调了早期诊断和多学科方法在LD管理中的重要性。疾病晚期的特点是具有重大的临床挑战,需要神经学家、癫痫学家和其他医疗保健专业人员之间的密切合作,并辅以有效的家庭护理。阿普利亚明显较高的患病率值得进一步调查潜在的遗传或环境因素。结论:本研究突出了LD的临床负担,强调了多学科管理的重要性,特别是在晚期。由专业团队和护理人员支持的家庭护理对于优化患者的健康至关重要。需要进一步的研究来确定早期生物标志物并开发针对这种破坏性疾病的靶向治疗方法。
Clinical course and management challenges in Lafora disease: a narrative analysis in an Apulian cohort.
Background: Lafora disease (LD) is an ultra-rare, autosomal recessive neurodegenerative disorder characterized by the accumulation of Lafora bodies in the brain, leading to drug-resistant epilepsy, myoclonus, progressive dementia, and cerebellar dysfunction. This retrospective study describes the clinical course and management challenges of LD in a cohort of patients from the Apulia region of Southern Italy, where the disease prevalence appears to be higher than in other populations.
Methods: We retrospectively analyzed clinical, electroencephalographic, and management data from six unrelated families with a confirmed diagnosis of LD, followed at the Neurology Unit of the Scientific Institute Casa Sollievo della Sofferenza Hospital between 2010 and 2024. Demographic information, clinical presentation, treatment history, disease progression, and outcomes were collected.
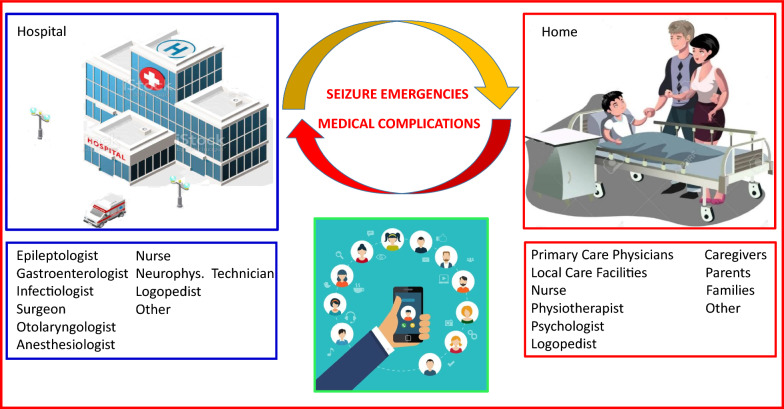
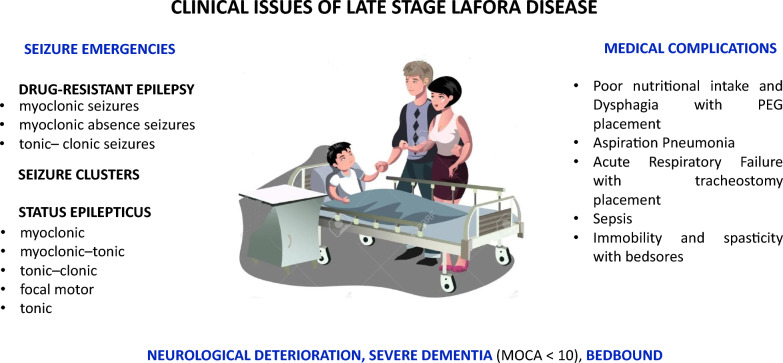
Results: Our analysis identified three distinct electroclinical stages: an initial Presenting Symptoms Stage with the onset of seizures and subsequent development of myoclonus; a Progressive Neurodegeneration Stage characterized by drug-resistant epilepsy, dementia, and ataxia; and a Terminal Stage marked by severe disability, frequent seizure emergencies, and medical complications. Management in the late stages proved particularly challenging, requiring a multidisciplinary approach to address refractory seizures, status epilepticus, and medical complications such as aspiration pneumonia and respiratory failure. Home-based care, with specialized team support, played a crucial role in minimizing hospitalizations.
Discussion: Our findings underscore the importance of early diagnosis and a multidisciplinary approach in the management of LD. The late stages of the disease are characterized by significant clinical challenges necessitating close collaboration among neurologists, epileptologists, and other healthcare professionals, supported by effective home-based care. The apparent higher prevalence in Apulia warrants further investigation into potential genetic or environmental factors.
Conclusion: This study highlights the significant clinical burden of LD and emphasizes the importance of multidisciplinary management, particularly in the advanced stages. Home-based care supported by specialized teams and caregivers is essential for optimizing patient well-being. Further research is needed to identify early biomarkers and develop targeted therapies for this devastating condition.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


