Fumin Xue, Chao An, Zhi Lei, Shijie Dong, Yaqiong Guo, Jiangshan Hou, Jing Yu, Yuesheng Wang
{"title":"A20单倍体功能不全的临床特征及遗传分析。","authors":"Fumin Xue, Chao An, Zhi Lei, Shijie Dong, Yaqiong Guo, Jiangshan Hou, Jing Yu, Yuesheng Wang","doi":"10.1186/s13023-025-04004-8","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>To described clinical and genetic characteristics of 4 patients presenting A20 haploinsufficiency (HA20) treated at Children's hospital affiliated to Zhengzhou university from 2015 to 2024.</p><p><strong>Methods: </strong>A retrospective analysis was conducted on the clinical data, genetic testing results, and treatment outcomes of four children with HA20 treated at the Children's hospital affiliated to Zhengzhou university from 2015 to 2024.</p><p><strong>Results: </strong>All four patients developed symptoms before the age of 1 year, presenting with recurrent fever and abdominal pain with diarrhea. Most common characteristics were hematochezia, bipolar aphthosis, arthritis, skin eruption in 50% of patients. Lab tests revealed elevated inflammatory markers; all patients had anemia. Imaging showed intestinal mucosal edema, hip/knee joint effusions, and lymphadenopathy in one case. Endoscopy revealed gastrointestinal aphthosis in 100% of cases. Genetic testing identified TNFAIP3 mutations in all four patients, including one novel whole-gene deletion (6q23.3chr6:136700000-138880000), 2 novel pathogenic mutations (c.866delA, c.1243_1247del), and one previously reported mutation (c.133C > T). Treatment included exclusive enteral nutrition (EEN) and thalidomide for all patients. One patient was switched to infliximab (IFX) combined with azathioprine due to gastrointestinal side effects, and one patient received methylprednisolone during acute phase. Follow-up for 4-10 years showed that 1 patient had improved symptoms with IFX and azathioprine but still had intermittent fever and perianal aphthosis; While one patient demonstrated poor response to EEN-thalidomide therapy requiring regimen change, the other responded well. One patient exhibited normalized gastrointestinal function following EEN-thalidomide therapy, yet still required repeated hospitalizations for recurrent infections with progressively prolonged inter-episode intervals.</p><p><strong>Conclusion: </strong>HA20 has strong clinical heterogeneity, and genetic testing is crucial for diagnosis and guiding treatment. Early diagnosis and individualized treatment can improve prognosis.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"457"},"PeriodicalIF":3.5000,"publicationDate":"2025-08-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12382130/pdf/","citationCount":"0","resultStr":"{\"title\":\"Clinical features and genetic analysis of A20 haploinsufficiency.\",\"authors\":\"Fumin Xue, Chao An, Zhi Lei, Shijie Dong, Yaqiong Guo, Jiangshan Hou, Jing Yu, Yuesheng Wang\",\"doi\":\"10.1186/s13023-025-04004-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Objective: </strong>To described clinical and genetic characteristics of 4 patients presenting A20 haploinsufficiency (HA20) treated at Children's hospital affiliated to Zhengzhou university from 2015 to 2024.</p><p><strong>Methods: </strong>A retrospective analysis was conducted on the clinical data, genetic testing results, and treatment outcomes of four children with HA20 treated at the Children's hospital affiliated to Zhengzhou university from 2015 to 2024.</p><p><strong>Results: </strong>All four patients developed symptoms before the age of 1 year, presenting with recurrent fever and abdominal pain with diarrhea. Most common characteristics were hematochezia, bipolar aphthosis, arthritis, skin eruption in 50% of patients. Lab tests revealed elevated inflammatory markers; all patients had anemia. Imaging showed intestinal mucosal edema, hip/knee joint effusions, and lymphadenopathy in one case. Endoscopy revealed gastrointestinal aphthosis in 100% of cases. Genetic testing identified TNFAIP3 mutations in all four patients, including one novel whole-gene deletion (6q23.3chr6:136700000-138880000), 2 novel pathogenic mutations (c.866delA, c.1243_1247del), and one previously reported mutation (c.133C > T). Treatment included exclusive enteral nutrition (EEN) and thalidomide for all patients. One patient was switched to infliximab (IFX) combined with azathioprine due to gastrointestinal side effects, and one patient received methylprednisolone during acute phase. Follow-up for 4-10 years showed that 1 patient had improved symptoms with IFX and azathioprine but still had intermittent fever and perianal aphthosis; While one patient demonstrated poor response to EEN-thalidomide therapy requiring regimen change, the other responded well. One patient exhibited normalized gastrointestinal function following EEN-thalidomide therapy, yet still required repeated hospitalizations for recurrent infections with progressively prolonged inter-episode intervals.</p><p><strong>Conclusion: </strong>HA20 has strong clinical heterogeneity, and genetic testing is crucial for diagnosis and guiding treatment. Early diagnosis and individualized treatment can improve prognosis.</p>\",\"PeriodicalId\":19651,\"journal\":{\"name\":\"Orphanet Journal of Rare Diseases\",\"volume\":\"20 1\",\"pages\":\"457\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2025-08-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12382130/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Orphanet Journal of Rare Diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13023-025-04004-8\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-04004-8","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Clinical features and genetic analysis of A20 haploinsufficiency.
Objective: To described clinical and genetic characteristics of 4 patients presenting A20 haploinsufficiency (HA20) treated at Children's hospital affiliated to Zhengzhou university from 2015 to 2024.
Methods: A retrospective analysis was conducted on the clinical data, genetic testing results, and treatment outcomes of four children with HA20 treated at the Children's hospital affiliated to Zhengzhou university from 2015 to 2024.
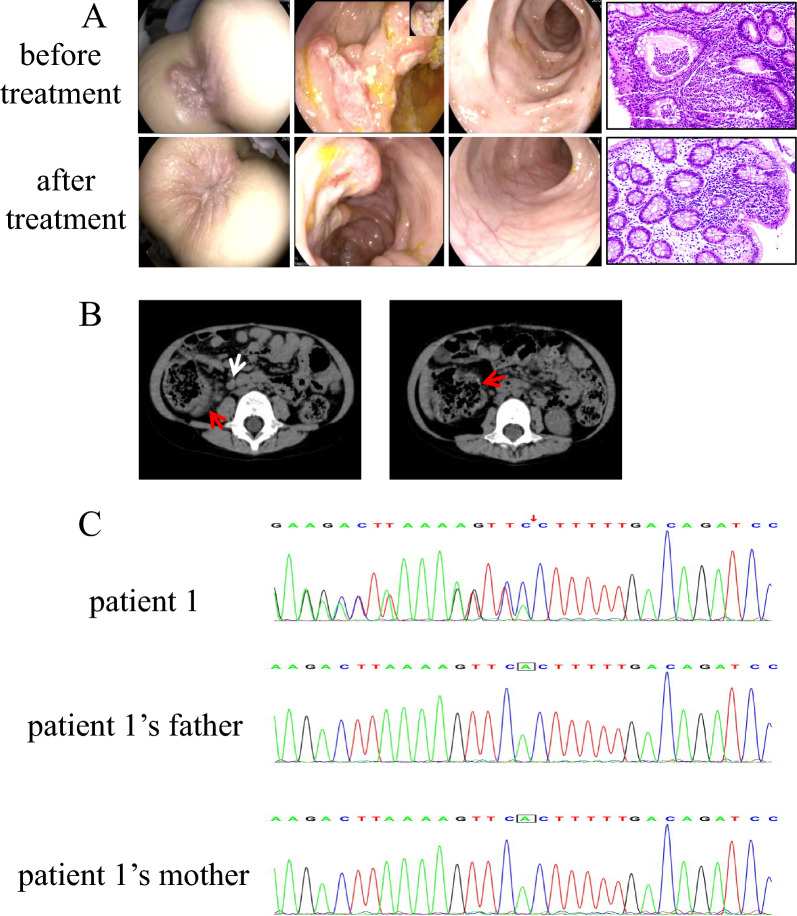
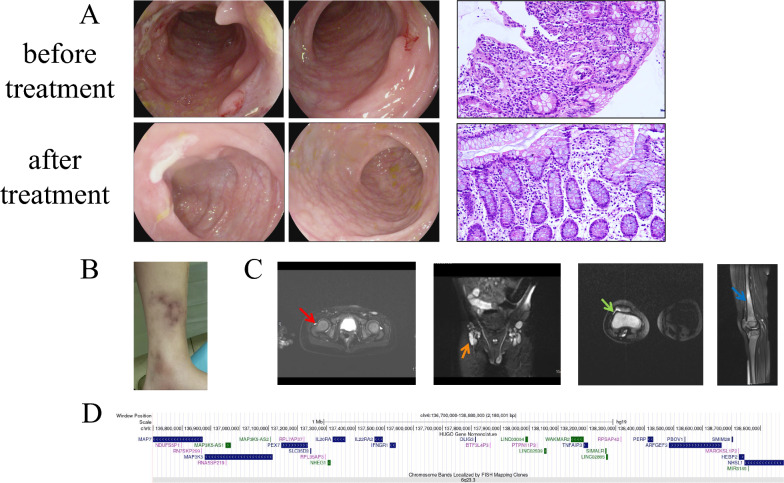
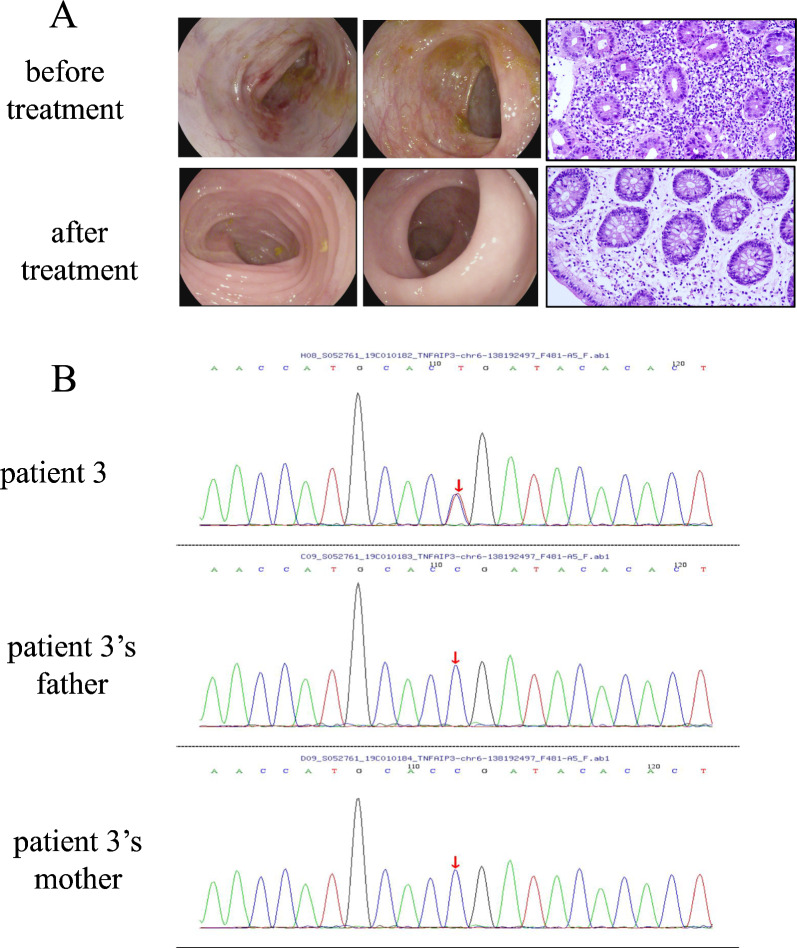
Results: All four patients developed symptoms before the age of 1 year, presenting with recurrent fever and abdominal pain with diarrhea. Most common characteristics were hematochezia, bipolar aphthosis, arthritis, skin eruption in 50% of patients. Lab tests revealed elevated inflammatory markers; all patients had anemia. Imaging showed intestinal mucosal edema, hip/knee joint effusions, and lymphadenopathy in one case. Endoscopy revealed gastrointestinal aphthosis in 100% of cases. Genetic testing identified TNFAIP3 mutations in all four patients, including one novel whole-gene deletion (6q23.3chr6:136700000-138880000), 2 novel pathogenic mutations (c.866delA, c.1243_1247del), and one previously reported mutation (c.133C > T). Treatment included exclusive enteral nutrition (EEN) and thalidomide for all patients. One patient was switched to infliximab (IFX) combined with azathioprine due to gastrointestinal side effects, and one patient received methylprednisolone during acute phase. Follow-up for 4-10 years showed that 1 patient had improved symptoms with IFX and azathioprine but still had intermittent fever and perianal aphthosis; While one patient demonstrated poor response to EEN-thalidomide therapy requiring regimen change, the other responded well. One patient exhibited normalized gastrointestinal function following EEN-thalidomide therapy, yet still required repeated hospitalizations for recurrent infections with progressively prolonged inter-episode intervals.
Conclusion: HA20 has strong clinical heterogeneity, and genetic testing is crucial for diagnosis and guiding treatment. Early diagnosis and individualized treatment can improve prognosis.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


