{"title":"FSHD1患者不常见特征的患病率和预测因素:来自法国FSHD登记的见解","authors":"Benoît Sanson, Abderhmane Slioui, Jérémy Garcia, Lori Klouvi, Julie Lejeune, Caroline Stalens, Céline Guien, Sitraka Rabarimeriarijaona, Rafaëlle Bernard, Juliette Nectoux, Sharham Attarian, Anne-Laure Bédat-Millet, Françoise Bouhour, François Constant Boyer, Jean-Baptiste Chanson, Ariane Choumert, Pascal Cintas, Elisa De La Cruz, Léonard Féasson, Maxime Fournier, Karima Ghorab, Agnès Jacquin-Piques, Pascal Laforêt, Armelle Magot, Maud Michaud, Jean-Baptiste Noury, Guilhem Solé, Marco Spinazzi, Tanya Stojkovic, Céline Tard, Luisa Villa, Christophe Béroud, Sabrina Sacconi","doi":"10.1186/s13023-025-03877-z","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Facioscapulohumeral muscular dystrophy (FSHD) is characterized by a typical pattern of muscle involvement, yet it encompasses a wide spectrum of phenotypes, including less common features that remain incompletely defined in the literature. While previous studies have highlighted this clinical variability, no consensus has been reached on how to classify uncommon manifestations, nor have specific predictors been identified. This study aims to describe these uncommon features and explore potential predictors, utilizing data from the French FSHD registry. To this end, we analysed data from 306 FSHD1 patients across nine French neuromuscular referral centres. Descriptive statistics, univariate analyses, and multiple logistic regression models were employed to examine uncommon characteristics and their predictors.</p><p><strong>Results: </strong>Uncommon features were observed in 19.6% of cases. The most common was a discrepancy between disease severity and D4Z4 repeat unit (RU) count (41.7%), followed by predominant impairment at proximal lower limb or distal upper limb muscles (21.7%). Three unanticipated features emerged: isolated or predominant axial impairment, anosmia and atopic dermatitis. Univariate analysis revealed that uncommon features were associated with higher RU count (6.5 ± 2.1 vs. 5.8 ± 1.8 in typical patients) and older age of onset (32.0 ± 18.8 years vs. 25.0 ± 15.4 years). Such features were more prevalent in the borderline 8-10 RU range, an association confirmed by multivariate analysis (OR = 2.43, 95% CI 1.21 to 4.87). Later age of onset consistently emerged as a factor across multiple multivariate models.</p><p><strong>Conclusions: </strong>This study documents uncommon FSHD features, revealing their association with the 8-10 RU range and later age of onset. These findings further support a complex interplay among genetic and epigenetic modifiers and ageing in shaping the clinical phenotype of FSHD, especially in patients carrying borderline D4Z4 arrays. Differential phenotypes, particularly in relation to RU range and age of onset, points to the importance of harmonized, comprehensive clinical and genetic assessments. Recognizing uncommon features may improve diagnostic accuracy and guide individualized management strategies, highlighting the need for tailored approaches to patient care.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"470"},"PeriodicalIF":3.5000,"publicationDate":"2025-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12403360/pdf/","citationCount":"0","resultStr":"{\"title\":\"Prevalence and predictors of uncommon features in FSHD1 patients: insights from the French FSHD registry.\",\"authors\":\"Benoît Sanson, Abderhmane Slioui, Jérémy Garcia, Lori Klouvi, Julie Lejeune, Caroline Stalens, Céline Guien, Sitraka Rabarimeriarijaona, Rafaëlle Bernard, Juliette Nectoux, Sharham Attarian, Anne-Laure Bédat-Millet, Françoise Bouhour, François Constant Boyer, Jean-Baptiste Chanson, Ariane Choumert, Pascal Cintas, Elisa De La Cruz, Léonard Féasson, Maxime Fournier, Karima Ghorab, Agnès Jacquin-Piques, Pascal Laforêt, Armelle Magot, Maud Michaud, Jean-Baptiste Noury, Guilhem Solé, Marco Spinazzi, Tanya Stojkovic, Céline Tard, Luisa Villa, Christophe Béroud, Sabrina Sacconi\",\"doi\":\"10.1186/s13023-025-03877-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Facioscapulohumeral muscular dystrophy (FSHD) is characterized by a typical pattern of muscle involvement, yet it encompasses a wide spectrum of phenotypes, including less common features that remain incompletely defined in the literature. While previous studies have highlighted this clinical variability, no consensus has been reached on how to classify uncommon manifestations, nor have specific predictors been identified. This study aims to describe these uncommon features and explore potential predictors, utilizing data from the French FSHD registry. To this end, we analysed data from 306 FSHD1 patients across nine French neuromuscular referral centres. Descriptive statistics, univariate analyses, and multiple logistic regression models were employed to examine uncommon characteristics and their predictors.</p><p><strong>Results: </strong>Uncommon features were observed in 19.6% of cases. The most common was a discrepancy between disease severity and D4Z4 repeat unit (RU) count (41.7%), followed by predominant impairment at proximal lower limb or distal upper limb muscles (21.7%). Three unanticipated features emerged: isolated or predominant axial impairment, anosmia and atopic dermatitis. Univariate analysis revealed that uncommon features were associated with higher RU count (6.5 ± 2.1 vs. 5.8 ± 1.8 in typical patients) and older age of onset (32.0 ± 18.8 years vs. 25.0 ± 15.4 years). Such features were more prevalent in the borderline 8-10 RU range, an association confirmed by multivariate analysis (OR = 2.43, 95% CI 1.21 to 4.87). Later age of onset consistently emerged as a factor across multiple multivariate models.</p><p><strong>Conclusions: </strong>This study documents uncommon FSHD features, revealing their association with the 8-10 RU range and later age of onset. These findings further support a complex interplay among genetic and epigenetic modifiers and ageing in shaping the clinical phenotype of FSHD, especially in patients carrying borderline D4Z4 arrays. Differential phenotypes, particularly in relation to RU range and age of onset, points to the importance of harmonized, comprehensive clinical and genetic assessments. Recognizing uncommon features may improve diagnostic accuracy and guide individualized management strategies, highlighting the need for tailored approaches to patient care.</p>\",\"PeriodicalId\":19651,\"journal\":{\"name\":\"Orphanet Journal of Rare Diseases\",\"volume\":\"20 1\",\"pages\":\"470\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2025-09-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12403360/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Orphanet Journal of Rare Diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13023-025-03877-z\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-03877-z","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
背景:面肩肱骨肌营养不良症(FSHD)以典型的肌肉受累模式为特征,但它包括广泛的表型,包括文献中尚未完全定义的不太常见的特征。虽然以前的研究强调了这种临床变异性,但对于如何分类罕见的表现没有达成共识,也没有确定具体的预测因素。本研究旨在描述这些不常见的特征,并利用法国FSHD登记处的数据探索潜在的预测因素。为此,我们分析了来自法国9个神经肌肉转诊中心的306名FSHD1患者的数据。采用描述性统计、单变量分析和多元逻辑回归模型来检验不常见特征及其预测因子。结果:19.6%的病例出现异常。最常见的是疾病严重程度和D4Z4重复单元(RU)计数之间的差异(41.7%),其次是下肢近端或上肢远端肌肉的主要损伤(21.7%)。出现了三个意想不到的特征:孤立的或主要的轴损伤,嗅觉丧失和特应性皮炎。单因素分析显示,不常见的特征与较高的RU计数(6.5±2.1 vs.典型患者的5.8±1.8)和较大的发病年龄(32.0±18.8岁vs. 25.0±15.4岁)相关。这些特征在8-10 RU的边界范围内更为普遍,多变量分析证实了这种关联(OR = 2.43, 95% CI 1.21至4.87)。在多个多变量模型中,较晚的发病年龄一直是一个因素。结论:本研究记录了罕见的FSHD特征,揭示了它们与8-10 RU范围和发病年龄的关系。这些发现进一步支持遗传和表观遗传修饰因子与衰老之间复杂的相互作用,形成FSHD的临床表型,特别是在携带边缘性D4Z4阵列的患者中。差异表型,特别是与RU范围和发病年龄相关的表型,指出了协调、全面的临床和遗传评估的重要性。认识到不寻常的特征可以提高诊断的准确性,并指导个性化的管理策略,强调需要量身定制的方法来治疗患者。
Prevalence and predictors of uncommon features in FSHD1 patients: insights from the French FSHD registry.
Background: Facioscapulohumeral muscular dystrophy (FSHD) is characterized by a typical pattern of muscle involvement, yet it encompasses a wide spectrum of phenotypes, including less common features that remain incompletely defined in the literature. While previous studies have highlighted this clinical variability, no consensus has been reached on how to classify uncommon manifestations, nor have specific predictors been identified. This study aims to describe these uncommon features and explore potential predictors, utilizing data from the French FSHD registry. To this end, we analysed data from 306 FSHD1 patients across nine French neuromuscular referral centres. Descriptive statistics, univariate analyses, and multiple logistic regression models were employed to examine uncommon characteristics and their predictors.
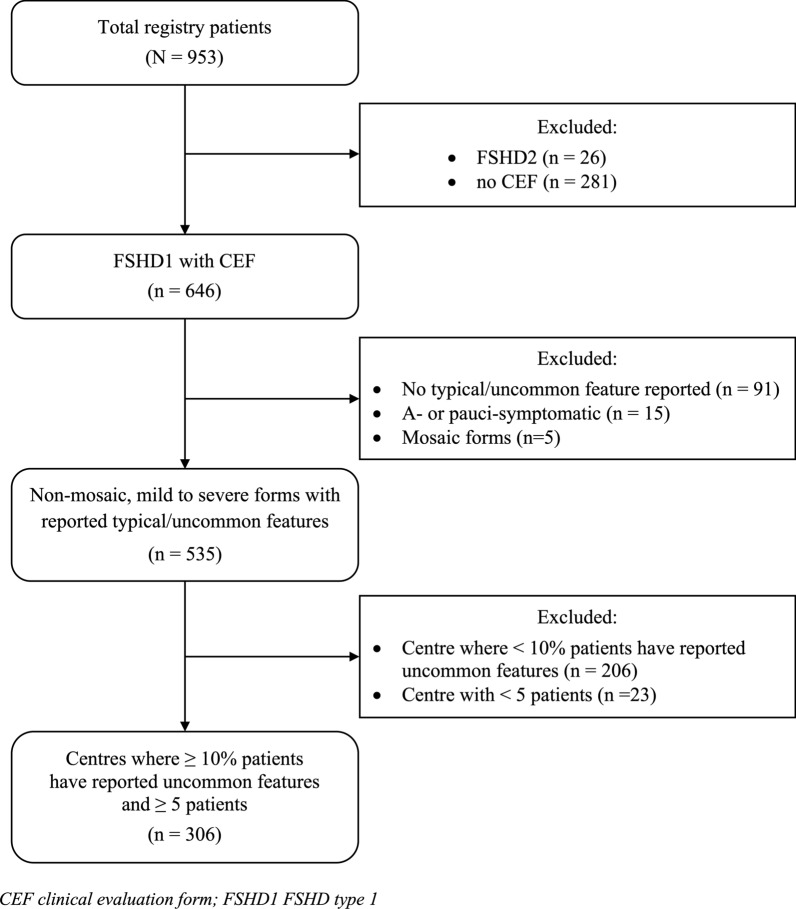
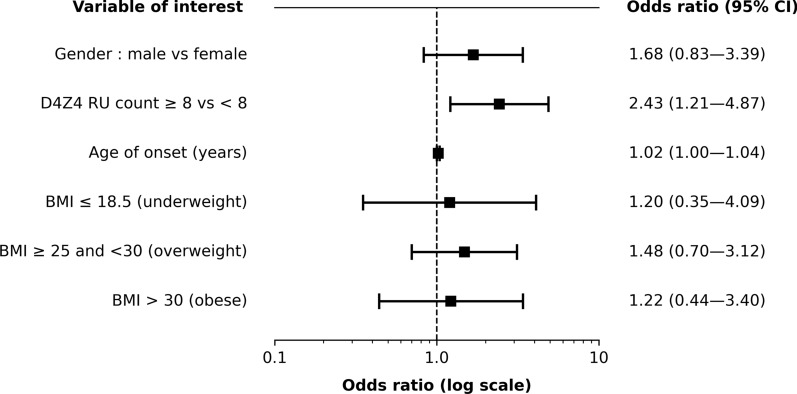
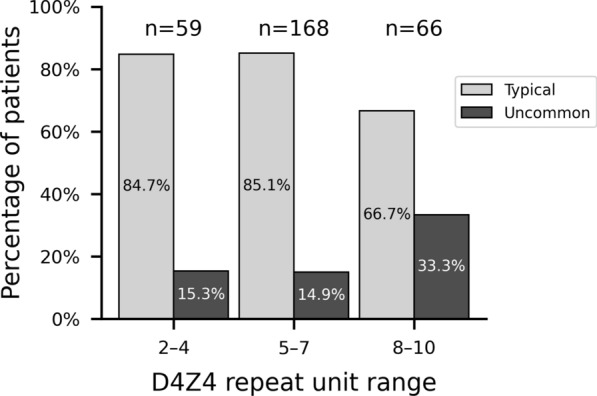
Results: Uncommon features were observed in 19.6% of cases. The most common was a discrepancy between disease severity and D4Z4 repeat unit (RU) count (41.7%), followed by predominant impairment at proximal lower limb or distal upper limb muscles (21.7%). Three unanticipated features emerged: isolated or predominant axial impairment, anosmia and atopic dermatitis. Univariate analysis revealed that uncommon features were associated with higher RU count (6.5 ± 2.1 vs. 5.8 ± 1.8 in typical patients) and older age of onset (32.0 ± 18.8 years vs. 25.0 ± 15.4 years). Such features were more prevalent in the borderline 8-10 RU range, an association confirmed by multivariate analysis (OR = 2.43, 95% CI 1.21 to 4.87). Later age of onset consistently emerged as a factor across multiple multivariate models.
Conclusions: This study documents uncommon FSHD features, revealing their association with the 8-10 RU range and later age of onset. These findings further support a complex interplay among genetic and epigenetic modifiers and ageing in shaping the clinical phenotype of FSHD, especially in patients carrying borderline D4Z4 arrays. Differential phenotypes, particularly in relation to RU range and age of onset, points to the importance of harmonized, comprehensive clinical and genetic assessments. Recognizing uncommon features may improve diagnostic accuracy and guide individualized management strategies, highlighting the need for tailored approaches to patient care.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


