{"title":"遗传性甲状腺转蛋白淀粉样变的遗传和临床特征:日本转诊中心十年的经验。","authors":"Toshiya Nomura, Yohei Misumi, Masayoshi Tasaki, Shiori Yamakawa, Tomoaki Taguchi, Konen Obayashi, Taro Yamashita, Yukio Ando, Mitsuharu Ueda","doi":"10.1186/s13023-025-04006-6","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Hereditary transthyretin (ATTRv) amyloidosis is a rare, intractable genetic disorder caused by mutations in the transthyretin (TTR) gene. More than 150 TTR mutations have been identified, along with genotype-phenotype correlations. Early diagnosis is critical to facilitate the timely initiation of disease-modifying therapies.</p><p><strong>Objective: </strong>To characterize the genetic and clinical features of ATTRv amyloidosis at our referral center.</p><p><strong>Methods: </strong>A total of 6,201 TTR genetic tests were conducted at Kumamoto University Hospital over a ten-year period and revealed 289 mutations, including 235 symptomatic cases, which were analyzed in this study.</p><p><strong>Results: </strong>In a cohort of 235 patients with symptomatic ATTRv amyloidosis, 46 TTR mutations were identified. The genotypes were distributed as follows: V30M in endemic areas (7.7%), V30M in non-endemic areas (48.5%), and non-V30M mutations (43.8%). The mean age of onset was lowest for patients with V30M in endemic areas (42.4 ± 15.6 years) and higher for those with non-V30M mutations (60.6 ± 14.6 years) and V30M in non-endemic areas (64.5 ± 11.9 years). Family history was present in 93.3% of V30M cases in endemic areas but absent in 42.0% of V30M cases in non-endemic areas and 57.5% of non-V30M cases. Polyneuropathy was the predominant initial symptom, affecting 73.7% of endemic V30M cases, 54.3% of non-endemic V30M cases, and 34.6% of non-V30M cases. Diagnosis occurred earlier in patients with V30M in endemic areas than in other groups. Notably, delayed diagnosis has been observed in patients presenting with carpal tunnel syndrome or polyneuropathy.</p><p><strong>Conclusions: </strong>These findings demonstrate that patients with V30M in non-endemic areas and those with non-V30M mutations are more prevalent than previously recognized and that their genetic and clinical characteristics exhibit considerable diversity.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"474"},"PeriodicalIF":3.5000,"publicationDate":"2025-09-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12406441/pdf/","citationCount":"0","resultStr":"{\"title\":\"Genetic and clinical features of hereditary transthyretin amyloidosis: a decade of experience at a Japanese referral center.\",\"authors\":\"Toshiya Nomura, Yohei Misumi, Masayoshi Tasaki, Shiori Yamakawa, Tomoaki Taguchi, Konen Obayashi, Taro Yamashita, Yukio Ando, Mitsuharu Ueda\",\"doi\":\"10.1186/s13023-025-04006-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Hereditary transthyretin (ATTRv) amyloidosis is a rare, intractable genetic disorder caused by mutations in the transthyretin (TTR) gene. More than 150 TTR mutations have been identified, along with genotype-phenotype correlations. Early diagnosis is critical to facilitate the timely initiation of disease-modifying therapies.</p><p><strong>Objective: </strong>To characterize the genetic and clinical features of ATTRv amyloidosis at our referral center.</p><p><strong>Methods: </strong>A total of 6,201 TTR genetic tests were conducted at Kumamoto University Hospital over a ten-year period and revealed 289 mutations, including 235 symptomatic cases, which were analyzed in this study.</p><p><strong>Results: </strong>In a cohort of 235 patients with symptomatic ATTRv amyloidosis, 46 TTR mutations were identified. The genotypes were distributed as follows: V30M in endemic areas (7.7%), V30M in non-endemic areas (48.5%), and non-V30M mutations (43.8%). The mean age of onset was lowest for patients with V30M in endemic areas (42.4 ± 15.6 years) and higher for those with non-V30M mutations (60.6 ± 14.6 years) and V30M in non-endemic areas (64.5 ± 11.9 years). Family history was present in 93.3% of V30M cases in endemic areas but absent in 42.0% of V30M cases in non-endemic areas and 57.5% of non-V30M cases. Polyneuropathy was the predominant initial symptom, affecting 73.7% of endemic V30M cases, 54.3% of non-endemic V30M cases, and 34.6% of non-V30M cases. Diagnosis occurred earlier in patients with V30M in endemic areas than in other groups. Notably, delayed diagnosis has been observed in patients presenting with carpal tunnel syndrome or polyneuropathy.</p><p><strong>Conclusions: </strong>These findings demonstrate that patients with V30M in non-endemic areas and those with non-V30M mutations are more prevalent than previously recognized and that their genetic and clinical characteristics exhibit considerable diversity.</p>\",\"PeriodicalId\":19651,\"journal\":{\"name\":\"Orphanet Journal of Rare Diseases\",\"volume\":\"20 1\",\"pages\":\"474\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2025-09-02\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12406441/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Orphanet Journal of Rare Diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13023-025-04006-6\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-04006-6","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Genetic and clinical features of hereditary transthyretin amyloidosis: a decade of experience at a Japanese referral center.
Background: Hereditary transthyretin (ATTRv) amyloidosis is a rare, intractable genetic disorder caused by mutations in the transthyretin (TTR) gene. More than 150 TTR mutations have been identified, along with genotype-phenotype correlations. Early diagnosis is critical to facilitate the timely initiation of disease-modifying therapies.
Objective: To characterize the genetic and clinical features of ATTRv amyloidosis at our referral center.
Methods: A total of 6,201 TTR genetic tests were conducted at Kumamoto University Hospital over a ten-year period and revealed 289 mutations, including 235 symptomatic cases, which were analyzed in this study.
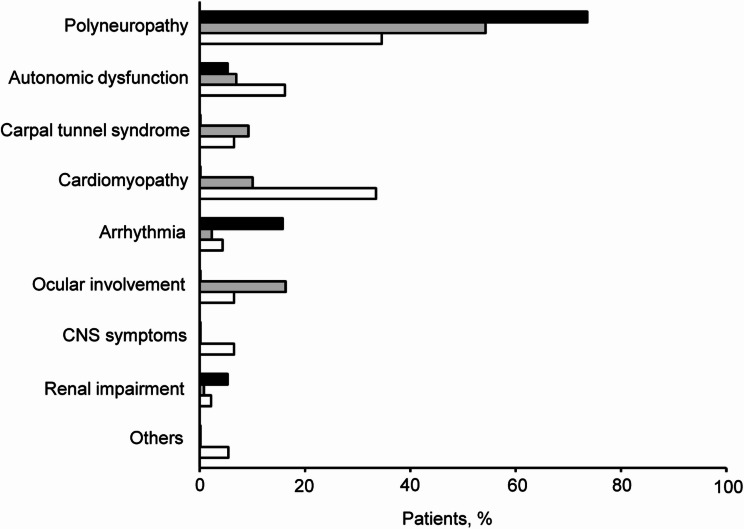
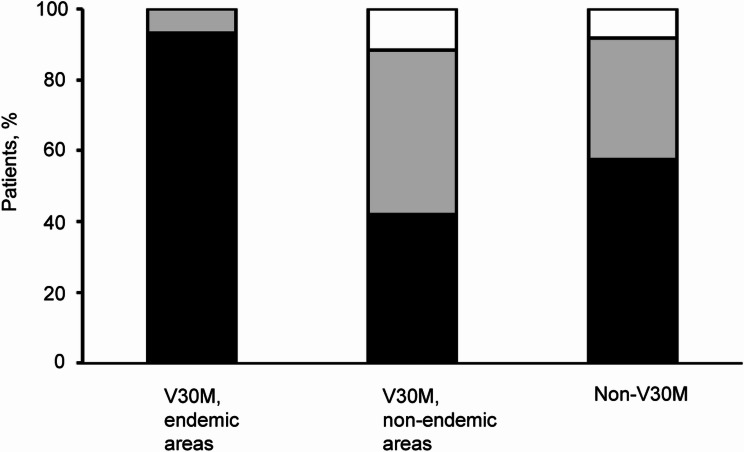
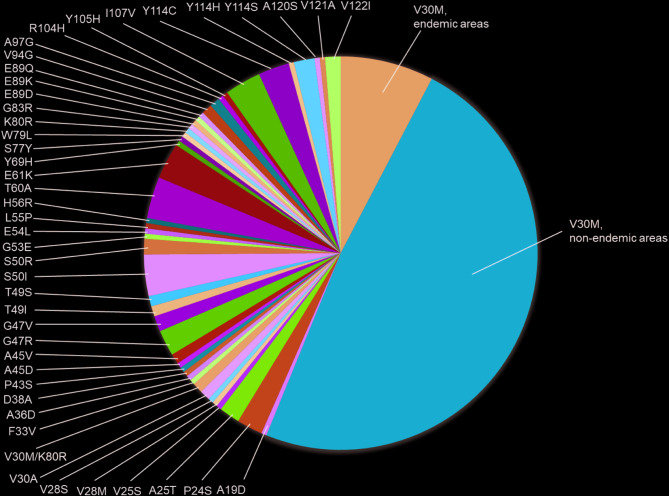
Results: In a cohort of 235 patients with symptomatic ATTRv amyloidosis, 46 TTR mutations were identified. The genotypes were distributed as follows: V30M in endemic areas (7.7%), V30M in non-endemic areas (48.5%), and non-V30M mutations (43.8%). The mean age of onset was lowest for patients with V30M in endemic areas (42.4 ± 15.6 years) and higher for those with non-V30M mutations (60.6 ± 14.6 years) and V30M in non-endemic areas (64.5 ± 11.9 years). Family history was present in 93.3% of V30M cases in endemic areas but absent in 42.0% of V30M cases in non-endemic areas and 57.5% of non-V30M cases. Polyneuropathy was the predominant initial symptom, affecting 73.7% of endemic V30M cases, 54.3% of non-endemic V30M cases, and 34.6% of non-V30M cases. Diagnosis occurred earlier in patients with V30M in endemic areas than in other groups. Notably, delayed diagnosis has been observed in patients presenting with carpal tunnel syndrome or polyneuropathy.
Conclusions: These findings demonstrate that patients with V30M in non-endemic areas and those with non-V30M mutations are more prevalent than previously recognized and that their genetic and clinical characteristics exhibit considerable diversity.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


