Heather L Glasgow, Ying Zheng, Jessica N Brazelton, Li Tang, Randall T Hayden
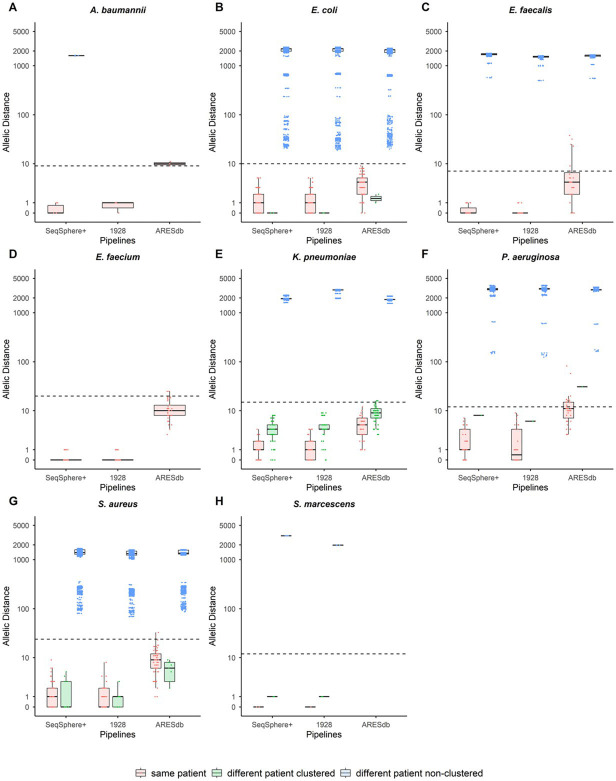
{"title":"医院暴发常见病原菌检测核心基因组多位点测序分型管道的比较。","authors":"Heather L Glasgow, Ying Zheng, Jessica N Brazelton, Li Tang, Randall T Hayden","doi":"10.1128/jcm.00646-25","DOIUrl":null,"url":null,"abstract":"<p><p>Microbial whole genome sequencing (WGS)-based methods have replaced conventional methods for genomic relatedness analysis in the investigation of or surveillance for infectious outbreaks. Analysis of WGS by core genome multi-locus sequence typing (cgMLST) has been proposed for standardized strain comparisons at high resolution and for longitudinal outbreak surveillance. We compared three commercial cgMLST software pipelines, Ridom SeqSphere+, 1928 Diagnostics' platform, and Ares Genetics ARESdb, for the identification of related (clustered) strains among 255 isolates of common bacterial pathogens, including <i>Acinetobacter baumannii, Escherichia coli, Enterococcus faecalis, Enterococcus faecium, Klebsiella pneumoniae, Pseudomonas aeruginosa, Staphylococcus aureus,</i> and <i>Serratia marcescens</i>. Isolates were previously identified as clustered with at least one other isolate collected from the same patient or different patients. Concordance with SeqSphere+ for differentiating clustered from non-clustered isolate pairs using suggested thresholds was 100% for the 1928 platform and 99.5% for ARESdb overall and 91.8%, 96.1%, and 100% among same-patient clustered, different-patient clustered, and different-patient non-clustered isolate pairs, respectively, in ARESdb. ARESdb showed significantly greater allelic distances than SeqSphere+ and 1928 among same-patient clustered isolate pairs (mean [standard deviation, SD], 7.6 [7.17], 1.18 [1.56], and 1 [1.59]) and different-patient clustered isolate pairs (mean [SD], 8.34 [4.31], 3.61 [2.26], and 3.91 [2.67]) (<i>P</i> < 0.0001), but not among non-clustered isolate pairs. For all species analyzed with sufficient sample size, allelic distances of clustered isolate pairs were significantly higher in ARESdb. CgMLST analysis using commercial pipelines may result in different allelic distances but showed concordance using suggested clustering thresholds to determine relatedness among strains.IMPORTANCEMicrobial genetic relatedness analysis is commonly used to investigate suspected outbreaks among different patients with infections caused by the same species of pathogen and, increasingly, for outbreak surveillance to uncover unsuspected healthcare-associated transmission events among patients, enabling early intervention by infection prevention and control specialists to prevent further spread. Here, we compared three commercial software tools for bacterial relatedness analysis, which perform gene-by-gene comparisons to determine the degree of relatedness for several species of common pathogens. Such software tools potentially allow clinical laboratories to perform rapid and routine analysis for infection control purposes without the need for in-house bioinformatic expertize. This study evaluates the comparability of three of these software tools, while presenting a model for comparative analytic pipeline evaluation.</p>","PeriodicalId":15511,"journal":{"name":"Journal of Clinical Microbiology","volume":" ","pages":"e0064625"},"PeriodicalIF":5.4000,"publicationDate":"2025-10-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12506026/pdf/","citationCount":"0","resultStr":"{\"title\":\"Comparison of core genome multi-locus sequencing typing pipelines for hospital outbreak detection of common bacterial pathogens.\",\"authors\":\"Heather L Glasgow, Ying Zheng, Jessica N Brazelton, Li Tang, Randall T Hayden\",\"doi\":\"10.1128/jcm.00646-25\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Microbial whole genome sequencing (WGS)-based methods have replaced conventional methods for genomic relatedness analysis in the investigation of or surveillance for infectious outbreaks. Analysis of WGS by core genome multi-locus sequence typing (cgMLST) has been proposed for standardized strain comparisons at high resolution and for longitudinal outbreak surveillance. We compared three commercial cgMLST software pipelines, Ridom SeqSphere+, 1928 Diagnostics' platform, and Ares Genetics ARESdb, for the identification of related (clustered) strains among 255 isolates of common bacterial pathogens, including <i>Acinetobacter baumannii, Escherichia coli, Enterococcus faecalis, Enterococcus faecium, Klebsiella pneumoniae, Pseudomonas aeruginosa, Staphylococcus aureus,</i> and <i>Serratia marcescens</i>. Isolates were previously identified as clustered with at least one other isolate collected from the same patient or different patients. Concordance with SeqSphere+ for differentiating clustered from non-clustered isolate pairs using suggested thresholds was 100% for the 1928 platform and 99.5% for ARESdb overall and 91.8%, 96.1%, and 100% among same-patient clustered, different-patient clustered, and different-patient non-clustered isolate pairs, respectively, in ARESdb. ARESdb showed significantly greater allelic distances than SeqSphere+ and 1928 among same-patient clustered isolate pairs (mean [standard deviation, SD], 7.6 [7.17], 1.18 [1.56], and 1 [1.59]) and different-patient clustered isolate pairs (mean [SD], 8.34 [4.31], 3.61 [2.26], and 3.91 [2.67]) (<i>P</i> < 0.0001), but not among non-clustered isolate pairs. For all species analyzed with sufficient sample size, allelic distances of clustered isolate pairs were significantly higher in ARESdb. CgMLST analysis using commercial pipelines may result in different allelic distances but showed concordance using suggested clustering thresholds to determine relatedness among strains.IMPORTANCEMicrobial genetic relatedness analysis is commonly used to investigate suspected outbreaks among different patients with infections caused by the same species of pathogen and, increasingly, for outbreak surveillance to uncover unsuspected healthcare-associated transmission events among patients, enabling early intervention by infection prevention and control specialists to prevent further spread. Here, we compared three commercial software tools for bacterial relatedness analysis, which perform gene-by-gene comparisons to determine the degree of relatedness for several species of common pathogens. Such software tools potentially allow clinical laboratories to perform rapid and routine analysis for infection control purposes without the need for in-house bioinformatic expertize. This study evaluates the comparability of three of these software tools, while presenting a model for comparative analytic pipeline evaluation.</p>\",\"PeriodicalId\":15511,\"journal\":{\"name\":\"Journal of Clinical Microbiology\",\"volume\":\" \",\"pages\":\"e0064625\"},\"PeriodicalIF\":5.4000,\"publicationDate\":\"2025-10-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12506026/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Clinical Microbiology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1128/jcm.00646-25\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/8/27 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Microbiology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1128/jcm.00646-25","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/8/27 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
Comparison of core genome multi-locus sequencing typing pipelines for hospital outbreak detection of common bacterial pathogens.
Microbial whole genome sequencing (WGS)-based methods have replaced conventional methods for genomic relatedness analysis in the investigation of or surveillance for infectious outbreaks. Analysis of WGS by core genome multi-locus sequence typing (cgMLST) has been proposed for standardized strain comparisons at high resolution and for longitudinal outbreak surveillance. We compared three commercial cgMLST software pipelines, Ridom SeqSphere+, 1928 Diagnostics' platform, and Ares Genetics ARESdb, for the identification of related (clustered) strains among 255 isolates of common bacterial pathogens, including Acinetobacter baumannii, Escherichia coli, Enterococcus faecalis, Enterococcus faecium, Klebsiella pneumoniae, Pseudomonas aeruginosa, Staphylococcus aureus, and Serratia marcescens. Isolates were previously identified as clustered with at least one other isolate collected from the same patient or different patients. Concordance with SeqSphere+ for differentiating clustered from non-clustered isolate pairs using suggested thresholds was 100% for the 1928 platform and 99.5% for ARESdb overall and 91.8%, 96.1%, and 100% among same-patient clustered, different-patient clustered, and different-patient non-clustered isolate pairs, respectively, in ARESdb. ARESdb showed significantly greater allelic distances than SeqSphere+ and 1928 among same-patient clustered isolate pairs (mean [standard deviation, SD], 7.6 [7.17], 1.18 [1.56], and 1 [1.59]) and different-patient clustered isolate pairs (mean [SD], 8.34 [4.31], 3.61 [2.26], and 3.91 [2.67]) (P < 0.0001), but not among non-clustered isolate pairs. For all species analyzed with sufficient sample size, allelic distances of clustered isolate pairs were significantly higher in ARESdb. CgMLST analysis using commercial pipelines may result in different allelic distances but showed concordance using suggested clustering thresholds to determine relatedness among strains.IMPORTANCEMicrobial genetic relatedness analysis is commonly used to investigate suspected outbreaks among different patients with infections caused by the same species of pathogen and, increasingly, for outbreak surveillance to uncover unsuspected healthcare-associated transmission events among patients, enabling early intervention by infection prevention and control specialists to prevent further spread. Here, we compared three commercial software tools for bacterial relatedness analysis, which perform gene-by-gene comparisons to determine the degree of relatedness for several species of common pathogens. Such software tools potentially allow clinical laboratories to perform rapid and routine analysis for infection control purposes without the need for in-house bioinformatic expertize. This study evaluates the comparability of three of these software tools, while presenting a model for comparative analytic pipeline evaluation.
期刊介绍:
The Journal of Clinical Microbiology® disseminates the latest research concerning the laboratory diagnosis of human and animal infections, along with the laboratory's role in epidemiology and the management of infectious diseases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


