Carmen Hoffbeck, Danielle M R L Middleton, Nicola J Nelson, Michael W Taylor
{"title":"基于Illumina和Oxford Nanopore扩增子测序的16S rRNA基因对标细菌分类分配方法","authors":"Carmen Hoffbeck, Danielle M R L Middleton, Nicola J Nelson, Michael W Taylor","doi":"10.1155/ijm/7563096","DOIUrl":null,"url":null,"abstract":"<p><p>Research investigating the microbial community of an ecosystem or animal can involve a range of methodologies, including sequencing technology, bioinformatic software and taxonomy database. Researchers may utilise short-read sequencing on Illumina MiSeq or long-read sequencing on platforms like Oxford Nanopore to obtain different research outcomes, for example, enhanced identification of microbes at species or strain level with Nanopore. However, replicability across these techniques is not well studied, while the technique used to process reads into microbial taxa may also result in different taxonomy assignments. In this study, we analyse an existing, real-world dataset which had low genus-level identification with Illumina sequencing and analysis with the SILVA database and compare sequencing with Nanopore on the same samples. We pair this with multiple bioinformatic approaches and taxonomy databases for each sequencing technique to compare phylum- and genus-level assignments and use mock communities to identify which combination of sequencing technique, bioinformatic approach and taxonomy database provides the most accurate taxonomy. We found that Nanopore reads processed with either utilised bioinformatic approach or taxonomy database provided higher accuracy in the assignment of a mock community than any technique combination with Illumina. We also found that the Top 10 genera assigned to a real-world database were substantially different across technique combinations and varied more by taxonomy database than either bioinformatic approach or sequencing technology.</p>","PeriodicalId":14098,"journal":{"name":"International Journal of Microbiology","volume":"2025 ","pages":"7563096"},"PeriodicalIF":3.2000,"publicationDate":"2025-08-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12367389/pdf/","citationCount":"0","resultStr":"{\"title\":\"Benchmarking 16S rRNA Gene-Based Approaches to Bacterial Taxonomy Assignment Based on Amplicon Sequencing With Illumina and Oxford Nanopore.\",\"authors\":\"Carmen Hoffbeck, Danielle M R L Middleton, Nicola J Nelson, Michael W Taylor\",\"doi\":\"10.1155/ijm/7563096\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Research investigating the microbial community of an ecosystem or animal can involve a range of methodologies, including sequencing technology, bioinformatic software and taxonomy database. Researchers may utilise short-read sequencing on Illumina MiSeq or long-read sequencing on platforms like Oxford Nanopore to obtain different research outcomes, for example, enhanced identification of microbes at species or strain level with Nanopore. However, replicability across these techniques is not well studied, while the technique used to process reads into microbial taxa may also result in different taxonomy assignments. In this study, we analyse an existing, real-world dataset which had low genus-level identification with Illumina sequencing and analysis with the SILVA database and compare sequencing with Nanopore on the same samples. We pair this with multiple bioinformatic approaches and taxonomy databases for each sequencing technique to compare phylum- and genus-level assignments and use mock communities to identify which combination of sequencing technique, bioinformatic approach and taxonomy database provides the most accurate taxonomy. We found that Nanopore reads processed with either utilised bioinformatic approach or taxonomy database provided higher accuracy in the assignment of a mock community than any technique combination with Illumina. We also found that the Top 10 genera assigned to a real-world database were substantially different across technique combinations and varied more by taxonomy database than either bioinformatic approach or sequencing technology.</p>\",\"PeriodicalId\":14098,\"journal\":{\"name\":\"International Journal of Microbiology\",\"volume\":\"2025 \",\"pages\":\"7563096\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2025-08-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12367389/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International Journal of Microbiology\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1155/ijm/7563096\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/1/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q3\",\"JCRName\":\"MICROBIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Microbiology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/ijm/7563096","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
Benchmarking 16S rRNA Gene-Based Approaches to Bacterial Taxonomy Assignment Based on Amplicon Sequencing With Illumina and Oxford Nanopore.
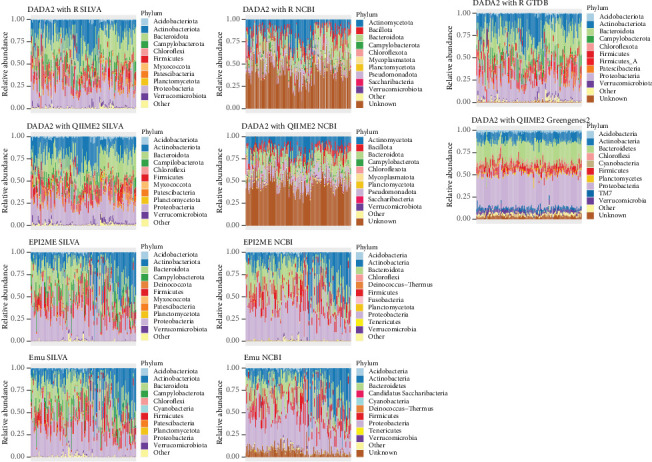
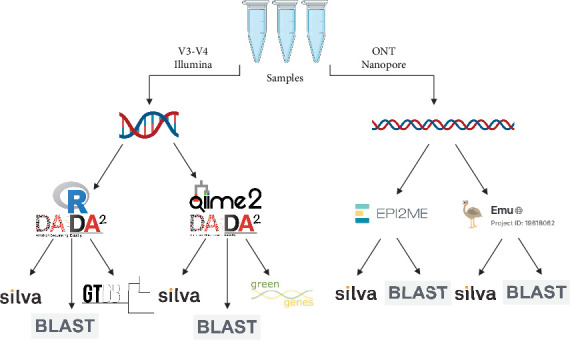
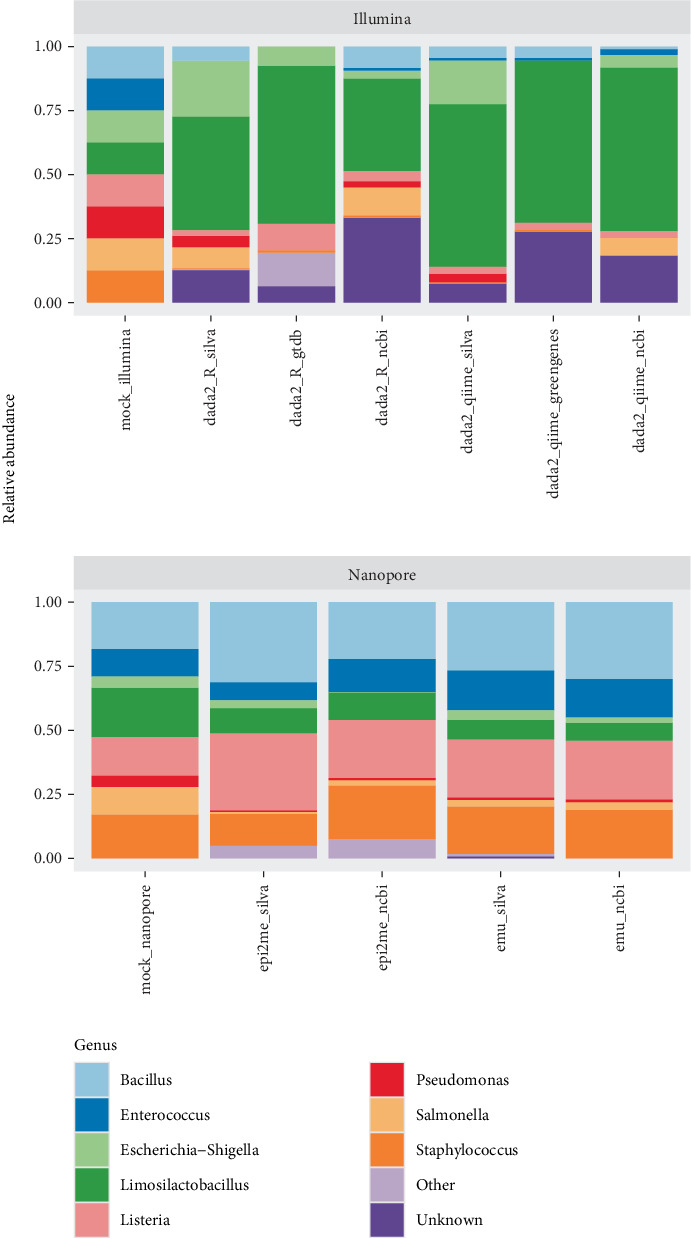
Research investigating the microbial community of an ecosystem or animal can involve a range of methodologies, including sequencing technology, bioinformatic software and taxonomy database. Researchers may utilise short-read sequencing on Illumina MiSeq or long-read sequencing on platforms like Oxford Nanopore to obtain different research outcomes, for example, enhanced identification of microbes at species or strain level with Nanopore. However, replicability across these techniques is not well studied, while the technique used to process reads into microbial taxa may also result in different taxonomy assignments. In this study, we analyse an existing, real-world dataset which had low genus-level identification with Illumina sequencing and analysis with the SILVA database and compare sequencing with Nanopore on the same samples. We pair this with multiple bioinformatic approaches and taxonomy databases for each sequencing technique to compare phylum- and genus-level assignments and use mock communities to identify which combination of sequencing technique, bioinformatic approach and taxonomy database provides the most accurate taxonomy. We found that Nanopore reads processed with either utilised bioinformatic approach or taxonomy database provided higher accuracy in the assignment of a mock community than any technique combination with Illumina. We also found that the Top 10 genera assigned to a real-world database were substantially different across technique combinations and varied more by taxonomy database than either bioinformatic approach or sequencing technology.
期刊介绍:
International Journal of Microbiology is a peer-reviewed, Open Access journal that publishes original research articles, review articles, and clinical studies on microorganisms and their interaction with hosts and the environment. The journal covers all microbes, including bacteria, fungi, viruses, archaea, and protozoa. Basic science will be considered, as well as medical and applied research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


