Max Gijsbertsen, Irene M J Mathijssen, Ana F Duarte Madancos, Johannes P T M van Leeuwen, Jeroen van de Peppel
{"title":"来自fgfr2相关综合征颅缝闭闭患者的三种人类诱导多能干细胞系的产生","authors":"Max Gijsbertsen, Irene M J Mathijssen, Ana F Duarte Madancos, Johannes P T M van Leeuwen, Jeroen van de Peppel","doi":"10.1242/dmm.052123","DOIUrl":null,"url":null,"abstract":"<p><p>Craniosynostosis is a multigenic congenital condition in which one or more calvarial sutures have prematurely fused during the development of the fetus. Pathogenic variants in FGFR2 are associated with the development of syndromic craniosynostosis, such as Crouzon, Apert and Pfeifer syndromes. Investigation of FGFR2-linked craniosynostosis is hindered by the lack of appropriate in vitro models. Patient-derived human induced pluripotent stem cell (hiPSC) in vitro disease models provide the opportunity to investigate the disease, identify molecular targets for pharmaceutical treatments, and enable the generation of autologous pluripotent stem cell catalogues. Here, we report three patient-derived hiPSC lines carrying the C342Y, S252W or E565G FGFR2 pathogenic variant. The patient hiPSC lines express characteristic pluripotency markers and display distinct phosphorylation profiles under unstimulated conditions. FGFR2C342Y showed autophosphorylation in the absence of bFGF ligand, although downstream docking proteins PLCγ and FRS2α were not phosphorylated. FGFR2S252W and FGFR2E565G hiPSCs showed increased phosphorylation of docking proteins PLCγ and FRS2α, whereas FGFR2 was not phosphorylated. These patient hiPSC lines provide molecular and cellular options to investigate FGFR2-linked craniosynostosis in the patient-specific genomic context and develop therapeutic modalities.</p>","PeriodicalId":11144,"journal":{"name":"Disease Models & Mechanisms","volume":" ","pages":""},"PeriodicalIF":3.3000,"publicationDate":"2025-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12486208/pdf/","citationCount":"0","resultStr":"{\"title\":\"Generation of human induced pluripotent stem cell lines from patients with FGFR2-linked syndromic craniosynostosis.\",\"authors\":\"Max Gijsbertsen, Irene M J Mathijssen, Ana F Duarte Madancos, Johannes P T M van Leeuwen, Jeroen van de Peppel\",\"doi\":\"10.1242/dmm.052123\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Craniosynostosis is a multigenic congenital condition in which one or more calvarial sutures have prematurely fused during the development of the fetus. Pathogenic variants in FGFR2 are associated with the development of syndromic craniosynostosis, such as Crouzon, Apert and Pfeifer syndromes. Investigation of FGFR2-linked craniosynostosis is hindered by the lack of appropriate in vitro models. Patient-derived human induced pluripotent stem cell (hiPSC) in vitro disease models provide the opportunity to investigate the disease, identify molecular targets for pharmaceutical treatments, and enable the generation of autologous pluripotent stem cell catalogues. Here, we report three patient-derived hiPSC lines carrying the C342Y, S252W or E565G FGFR2 pathogenic variant. The patient hiPSC lines express characteristic pluripotency markers and display distinct phosphorylation profiles under unstimulated conditions. FGFR2C342Y showed autophosphorylation in the absence of bFGF ligand, although downstream docking proteins PLCγ and FRS2α were not phosphorylated. FGFR2S252W and FGFR2E565G hiPSCs showed increased phosphorylation of docking proteins PLCγ and FRS2α, whereas FGFR2 was not phosphorylated. These patient hiPSC lines provide molecular and cellular options to investigate FGFR2-linked craniosynostosis in the patient-specific genomic context and develop therapeutic modalities.</p>\",\"PeriodicalId\":11144,\"journal\":{\"name\":\"Disease Models & Mechanisms\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2025-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12486208/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Disease Models & Mechanisms\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1242/dmm.052123\",\"RegionNum\":3,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/9/18 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Disease Models & Mechanisms","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1242/dmm.052123","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/9/18 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
Generation of human induced pluripotent stem cell lines from patients with FGFR2-linked syndromic craniosynostosis.
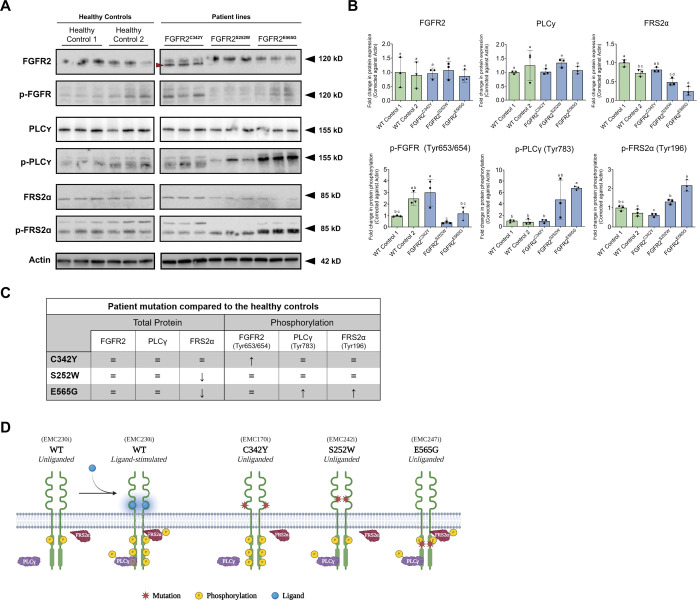
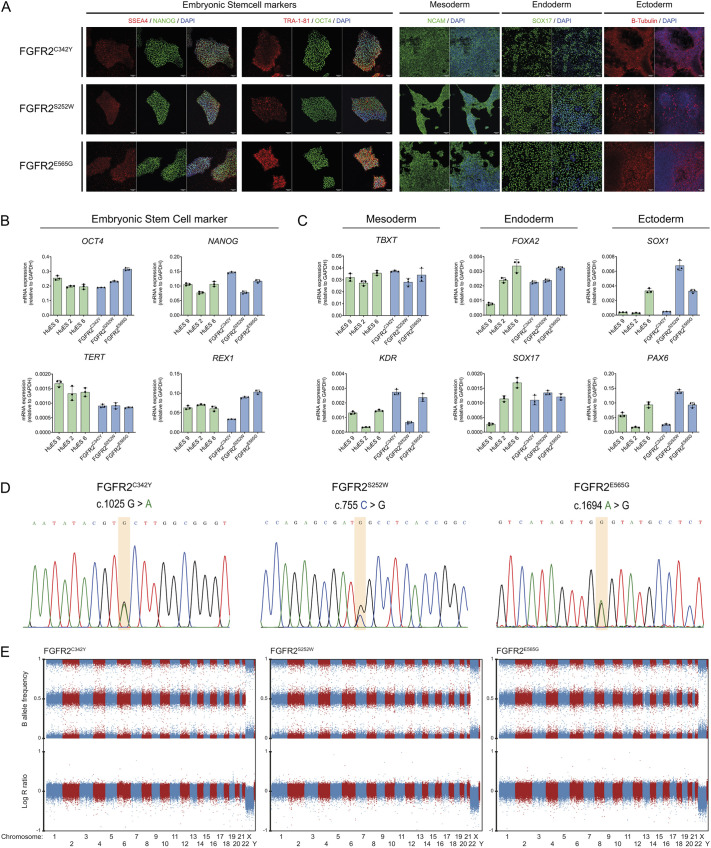
Craniosynostosis is a multigenic congenital condition in which one or more calvarial sutures have prematurely fused during the development of the fetus. Pathogenic variants in FGFR2 are associated with the development of syndromic craniosynostosis, such as Crouzon, Apert and Pfeifer syndromes. Investigation of FGFR2-linked craniosynostosis is hindered by the lack of appropriate in vitro models. Patient-derived human induced pluripotent stem cell (hiPSC) in vitro disease models provide the opportunity to investigate the disease, identify molecular targets for pharmaceutical treatments, and enable the generation of autologous pluripotent stem cell catalogues. Here, we report three patient-derived hiPSC lines carrying the C342Y, S252W or E565G FGFR2 pathogenic variant. The patient hiPSC lines express characteristic pluripotency markers and display distinct phosphorylation profiles under unstimulated conditions. FGFR2C342Y showed autophosphorylation in the absence of bFGF ligand, although downstream docking proteins PLCγ and FRS2α were not phosphorylated. FGFR2S252W and FGFR2E565G hiPSCs showed increased phosphorylation of docking proteins PLCγ and FRS2α, whereas FGFR2 was not phosphorylated. These patient hiPSC lines provide molecular and cellular options to investigate FGFR2-linked craniosynostosis in the patient-specific genomic context and develop therapeutic modalities.
期刊介绍:
Disease Models & Mechanisms (DMM) is an online Open Access journal focusing on the use of model systems to better understand, diagnose and treat human disease.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


