Giorgia Mandrile, Gill Rumsby, Veronica Sciannameo, Andrea G Cogal, Michelle Glover, John C Lieske, Peter C Harris
{"title":"原发性高草酸尿的全球遗传患病率估计比以前报道的要高。","authors":"Giorgia Mandrile, Gill Rumsby, Veronica Sciannameo, Andrea G Cogal, Michelle Glover, John C Lieske, Peter C Harris","doi":"10.1093/ckj/sfaf194","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Primary hyperoxaluria (PH), a rare autosomal recessive disease of oxalate accumulation in the kidneys, is caused by biallelic pathogenic changes in three known genes: <i>AGXT</i> (PH1), <i>GRHPR</i> (PH2) and <i>HOGA1</i> (PH3).</p><p><strong>Methods: </strong>To better understand the overall risk of developing clinical PH, we manually curated and classified PH genetic variants and calculated the estimated genetic prevalence overall and in five ethnic subpopulations using allelic frequencies from the population Genome Aggregation Database (gnomAD version 2.1.1).</p><p><strong>Results: </strong>Of the 651 identified PH variants, 273 were found in gnomAD 2.1.1 on the day of download and after reclassification, 208 were determined pathogenic (P) or likely pathogenic (LP) (<i>AGXT, n</i> = 94; <i>GRHPR, n</i> = 46; and <i>HOGA1, n</i> = 68) and a further 65 were classified as rare variants of uncertain significance (VUS). Using P and LP only, estimated carrier frequency was 1:229 for PH1, 1:465 for PH2 and 1:151 for PH3, while genetic prevalence was 1:209 357 for PH1, 1:863 028 for PH2 and 1:90 834 for PH3 (i.e. nearly 5, 1 and 11 per 1 million individuals, respectively). The highest carrier frequencies for <i>AGXT</i> pathogenic variants were in East Asians (1 in 146) and the European non-Finnish population (1 in 187); for <i>GRHPR</i>, South Asians (1 in 313) and the European non-Finnish population (1 in 413); and for <i>HOGA1</i>, Ashkenazi Jewish (1 in 38) and East Asians (1 in 100). The estimated risk of developing PH was ≈1:59 017.</p><p><strong>Conclusions: </strong>This careful benchmarking exercise indicates that a significant number of individuals at risk for PH symptoms remain undiagnosed. Since these numbers exceed known diagnosed cases of PH, improved screening and diagnosis of this underestimated disease is necessary.</p>","PeriodicalId":10435,"journal":{"name":"Clinical Kidney Journal","volume":"18 7","pages":"sfaf194"},"PeriodicalIF":4.6000,"publicationDate":"2025-06-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12378434/pdf/","citationCount":"0","resultStr":"{\"title\":\"Global genetic prevalence estimates of primary hyperoxaluria are greater than previously reported.\",\"authors\":\"Giorgia Mandrile, Gill Rumsby, Veronica Sciannameo, Andrea G Cogal, Michelle Glover, John C Lieske, Peter C Harris\",\"doi\":\"10.1093/ckj/sfaf194\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Primary hyperoxaluria (PH), a rare autosomal recessive disease of oxalate accumulation in the kidneys, is caused by biallelic pathogenic changes in three known genes: <i>AGXT</i> (PH1), <i>GRHPR</i> (PH2) and <i>HOGA1</i> (PH3).</p><p><strong>Methods: </strong>To better understand the overall risk of developing clinical PH, we manually curated and classified PH genetic variants and calculated the estimated genetic prevalence overall and in five ethnic subpopulations using allelic frequencies from the population Genome Aggregation Database (gnomAD version 2.1.1).</p><p><strong>Results: </strong>Of the 651 identified PH variants, 273 were found in gnomAD 2.1.1 on the day of download and after reclassification, 208 were determined pathogenic (P) or likely pathogenic (LP) (<i>AGXT, n</i> = 94; <i>GRHPR, n</i> = 46; and <i>HOGA1, n</i> = 68) and a further 65 were classified as rare variants of uncertain significance (VUS). Using P and LP only, estimated carrier frequency was 1:229 for PH1, 1:465 for PH2 and 1:151 for PH3, while genetic prevalence was 1:209 357 for PH1, 1:863 028 for PH2 and 1:90 834 for PH3 (i.e. nearly 5, 1 and 11 per 1 million individuals, respectively). The highest carrier frequencies for <i>AGXT</i> pathogenic variants were in East Asians (1 in 146) and the European non-Finnish population (1 in 187); for <i>GRHPR</i>, South Asians (1 in 313) and the European non-Finnish population (1 in 413); and for <i>HOGA1</i>, Ashkenazi Jewish (1 in 38) and East Asians (1 in 100). The estimated risk of developing PH was ≈1:59 017.</p><p><strong>Conclusions: </strong>This careful benchmarking exercise indicates that a significant number of individuals at risk for PH symptoms remain undiagnosed. Since these numbers exceed known diagnosed cases of PH, improved screening and diagnosis of this underestimated disease is necessary.</p>\",\"PeriodicalId\":10435,\"journal\":{\"name\":\"Clinical Kidney Journal\",\"volume\":\"18 7\",\"pages\":\"sfaf194\"},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2025-06-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12378434/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Clinical Kidney Journal\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1093/ckj/sfaf194\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2025/7/1 0:00:00\",\"PubModel\":\"eCollection\",\"JCR\":\"Q1\",\"JCRName\":\"UROLOGY & NEPHROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Kidney Journal","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1093/ckj/sfaf194","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/7/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"UROLOGY & NEPHROLOGY","Score":null,"Total":0}
Global genetic prevalence estimates of primary hyperoxaluria are greater than previously reported.
Background: Primary hyperoxaluria (PH), a rare autosomal recessive disease of oxalate accumulation in the kidneys, is caused by biallelic pathogenic changes in three known genes: AGXT (PH1), GRHPR (PH2) and HOGA1 (PH3).
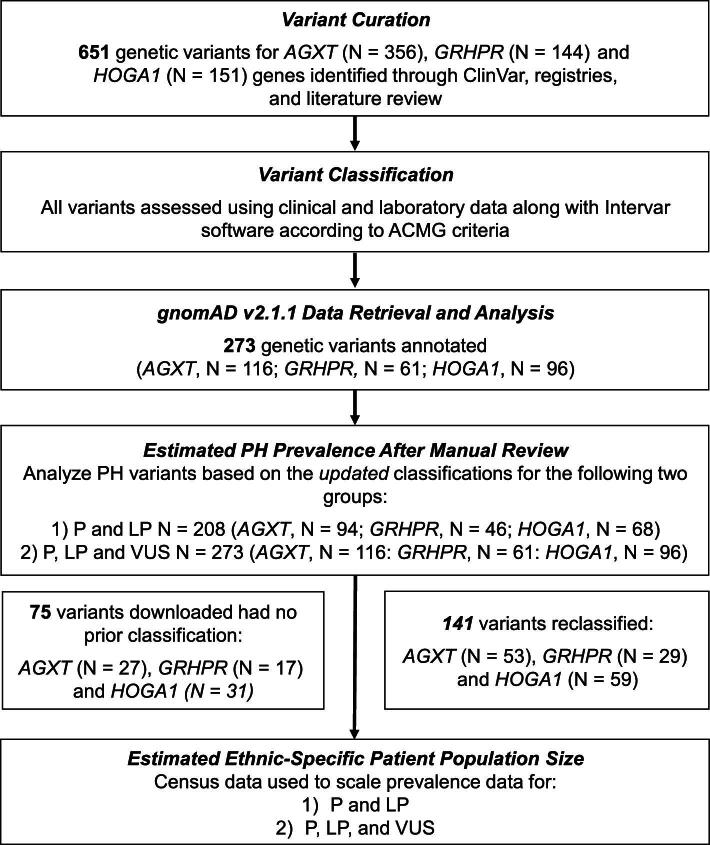
Methods: To better understand the overall risk of developing clinical PH, we manually curated and classified PH genetic variants and calculated the estimated genetic prevalence overall and in five ethnic subpopulations using allelic frequencies from the population Genome Aggregation Database (gnomAD version 2.1.1).
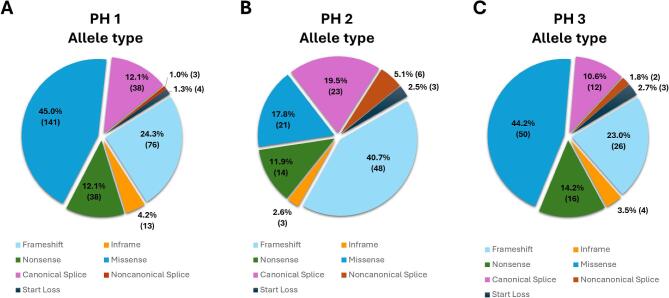
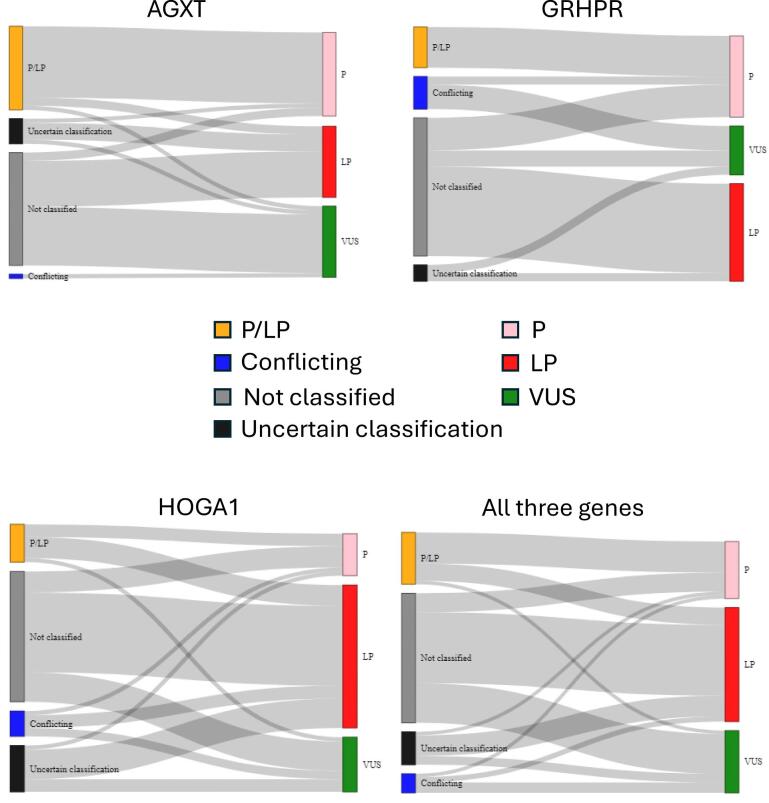
Results: Of the 651 identified PH variants, 273 were found in gnomAD 2.1.1 on the day of download and after reclassification, 208 were determined pathogenic (P) or likely pathogenic (LP) (AGXT, n = 94; GRHPR, n = 46; and HOGA1, n = 68) and a further 65 were classified as rare variants of uncertain significance (VUS). Using P and LP only, estimated carrier frequency was 1:229 for PH1, 1:465 for PH2 and 1:151 for PH3, while genetic prevalence was 1:209 357 for PH1, 1:863 028 for PH2 and 1:90 834 for PH3 (i.e. nearly 5, 1 and 11 per 1 million individuals, respectively). The highest carrier frequencies for AGXT pathogenic variants were in East Asians (1 in 146) and the European non-Finnish population (1 in 187); for GRHPR, South Asians (1 in 313) and the European non-Finnish population (1 in 413); and for HOGA1, Ashkenazi Jewish (1 in 38) and East Asians (1 in 100). The estimated risk of developing PH was ≈1:59 017.
Conclusions: This careful benchmarking exercise indicates that a significant number of individuals at risk for PH symptoms remain undiagnosed. Since these numbers exceed known diagnosed cases of PH, improved screening and diagnosis of this underestimated disease is necessary.
期刊介绍:
About the Journal
Clinical Kidney Journal: Clinical and Translational Nephrology (ckj), an official journal of the ERA-EDTA (European Renal Association-European Dialysis and Transplant Association), is a fully open access, online only journal publishing bimonthly. The journal is an essential educational and training resource integrating clinical, translational and educational research into clinical practice. ckj aims to contribute to a translational research culture among nephrologists and kidney pathologists that helps close the gap between basic researchers and practicing clinicians and promote sorely needed innovation in the Nephrology field. All research articles in this journal have undergone peer review.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


