{"title":"COL4A3基因致病性同义变异导致幼儿Alport综合征伴IgA沉积:1例报告。","authors":"Panpan Shao, Wenpei Liang, Rongrong Xv, Yonghua He, Jinbo Xiang, Xueqing Ma, Jinyun Pu, Jianhua Zhou, Huiqing Yuan, Liru Qiu","doi":"10.1186/s12882-025-04416-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Alport syndrome (AS) is a hereditary kidney disorder caused by pathogenic variations in COL4 genes and is clinically characterized by hematuria, proteinuria, and progressive renal impairment. IgA nephropathy (IgAN) is a clinicopathological syndrome characterized by the deposition of IgA or IgA-dominant in the glomerular mesangial areas.</p><p><strong>Case presentation: </strong>This article reports a case of a 2-year-and-3-month-old female toddler who presented with hematuria and proteinuria. Renal biopsy revealed IgA deposition, and a few segments displayed atypical tearing and layering changes in the dense layer. Family screening revealed that the father and grandmother of the patient had been diagnosed with thin basement membrane disease. Genetic testing revealed compound heterozygous variations c.4793T > G (p.Leu1598Arg) and c.765G > A (p.Thr255Thr) in the COL4A3 gene. Both hematuria and proteinuria improved significantly with treatment involving steroids, mycophenolate mofetil, tacrolimus, and angiotensin-converting enzyme inhibitors (ACEIs), but both recurred and slowly increased under ACEIs monotherapy. The toddler was ultimately diagnosed with AS comorbid with IgAN, and the variant c.765G > A (p.Thr255Thr) from the father is suspected to be pathogenic based on familial segregation and predictive evidence.</p><p><strong>Conclusion: </strong>Younger children with AS exhibit milder clinical manifestations or are asymptomatic. Biallelic pathogenic variations and IgA deposition may accelerate AS disease progression. Synonymous variations can also be pathogenic.</p>","PeriodicalId":9089,"journal":{"name":"BMC Nephrology","volume":"26 1","pages":"493"},"PeriodicalIF":2.4000,"publicationDate":"2025-08-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12382248/pdf/","citationCount":"0","resultStr":"{\"title\":\"Pathogenic synonymous variation of the COL4A3 gene causing Alport syndrome comorbid with IgA deposition in a toddler: a case report.\",\"authors\":\"Panpan Shao, Wenpei Liang, Rongrong Xv, Yonghua He, Jinbo Xiang, Xueqing Ma, Jinyun Pu, Jianhua Zhou, Huiqing Yuan, Liru Qiu\",\"doi\":\"10.1186/s12882-025-04416-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Alport syndrome (AS) is a hereditary kidney disorder caused by pathogenic variations in COL4 genes and is clinically characterized by hematuria, proteinuria, and progressive renal impairment. IgA nephropathy (IgAN) is a clinicopathological syndrome characterized by the deposition of IgA or IgA-dominant in the glomerular mesangial areas.</p><p><strong>Case presentation: </strong>This article reports a case of a 2-year-and-3-month-old female toddler who presented with hematuria and proteinuria. Renal biopsy revealed IgA deposition, and a few segments displayed atypical tearing and layering changes in the dense layer. Family screening revealed that the father and grandmother of the patient had been diagnosed with thin basement membrane disease. Genetic testing revealed compound heterozygous variations c.4793T > G (p.Leu1598Arg) and c.765G > A (p.Thr255Thr) in the COL4A3 gene. Both hematuria and proteinuria improved significantly with treatment involving steroids, mycophenolate mofetil, tacrolimus, and angiotensin-converting enzyme inhibitors (ACEIs), but both recurred and slowly increased under ACEIs monotherapy. The toddler was ultimately diagnosed with AS comorbid with IgAN, and the variant c.765G > A (p.Thr255Thr) from the father is suspected to be pathogenic based on familial segregation and predictive evidence.</p><p><strong>Conclusion: </strong>Younger children with AS exhibit milder clinical manifestations or are asymptomatic. Biallelic pathogenic variations and IgA deposition may accelerate AS disease progression. Synonymous variations can also be pathogenic.</p>\",\"PeriodicalId\":9089,\"journal\":{\"name\":\"BMC Nephrology\",\"volume\":\"26 1\",\"pages\":\"493\"},\"PeriodicalIF\":2.4000,\"publicationDate\":\"2025-08-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12382248/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"BMC Nephrology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s12882-025-04416-5\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"UROLOGY & NEPHROLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"BMC Nephrology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s12882-025-04416-5","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"UROLOGY & NEPHROLOGY","Score":null,"Total":0}
引用次数: 0
摘要
背景:Alport综合征(AS)是一种由COL4基因致病性变异引起的遗传性肾脏疾病,临床表现为血尿、蛋白尿和进行性肾损害。IgA肾病(IgAN)是一种以肾小球系膜区IgA或IgA显性沉积为特征的临床病理综合征。病例介绍:这篇文章报告了一个2岁3个月大的女婴谁提出血尿和蛋白尿。肾活检示IgA沉积,少数节段致密层不典型撕裂及分层改变。家庭筛查显示患者的父亲和祖母被诊断为基底膜薄病。基因检测发现COL4A3基因存在复合杂合变异c.4793T > G (p.l u1598arg)和c.765G > A (p.s r255thr)。血尿和蛋白尿在类固醇、霉酚酸酯、他克莫司和血管紧张素转换酶抑制剂(ACEIs)治疗后均显著改善,但在ACEIs单药治疗下,两者均复发并缓慢增加。该幼儿最终被诊断为AS与IgAN共病,根据家族分离和预测证据,来自父亲的c.765G >a (p.Thr255Thr)变异被怀疑具有致病性。结论:低龄AS患儿临床表现较轻或无症状。双等位基因致病变异和IgA沉积可能加速AS疾病的进展。同义变异也可能是致病的。
Pathogenic synonymous variation of the COL4A3 gene causing Alport syndrome comorbid with IgA deposition in a toddler: a case report.
Background: Alport syndrome (AS) is a hereditary kidney disorder caused by pathogenic variations in COL4 genes and is clinically characterized by hematuria, proteinuria, and progressive renal impairment. IgA nephropathy (IgAN) is a clinicopathological syndrome characterized by the deposition of IgA or IgA-dominant in the glomerular mesangial areas.
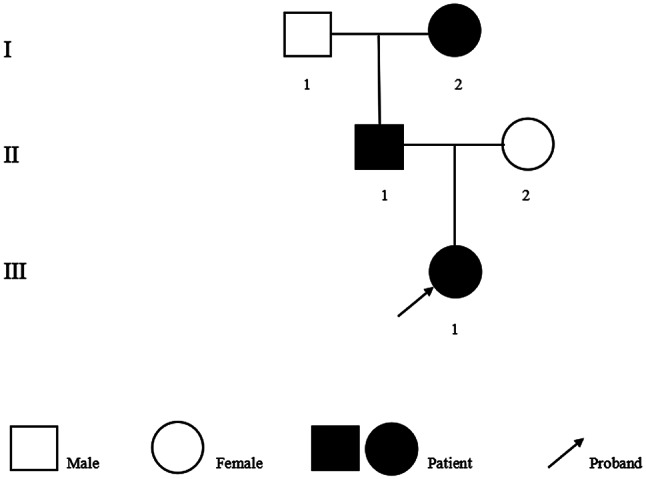
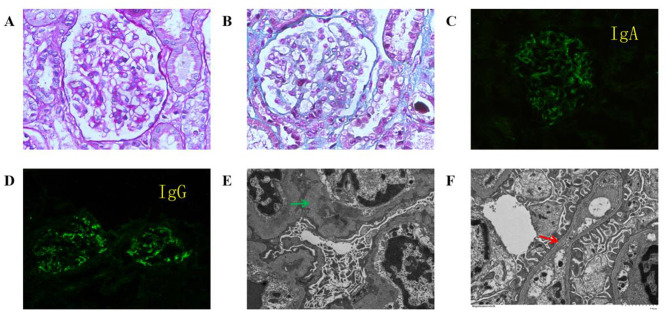
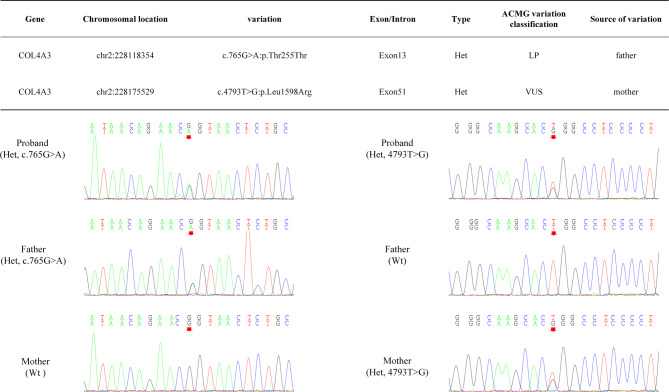
Case presentation: This article reports a case of a 2-year-and-3-month-old female toddler who presented with hematuria and proteinuria. Renal biopsy revealed IgA deposition, and a few segments displayed atypical tearing and layering changes in the dense layer. Family screening revealed that the father and grandmother of the patient had been diagnosed with thin basement membrane disease. Genetic testing revealed compound heterozygous variations c.4793T > G (p.Leu1598Arg) and c.765G > A (p.Thr255Thr) in the COL4A3 gene. Both hematuria and proteinuria improved significantly with treatment involving steroids, mycophenolate mofetil, tacrolimus, and angiotensin-converting enzyme inhibitors (ACEIs), but both recurred and slowly increased under ACEIs monotherapy. The toddler was ultimately diagnosed with AS comorbid with IgAN, and the variant c.765G > A (p.Thr255Thr) from the father is suspected to be pathogenic based on familial segregation and predictive evidence.
Conclusion: Younger children with AS exhibit milder clinical manifestations or are asymptomatic. Biallelic pathogenic variations and IgA deposition may accelerate AS disease progression. Synonymous variations can also be pathogenic.
期刊介绍:
BMC Nephrology is an open access journal publishing original peer-reviewed research articles in all aspects of the prevention, diagnosis and management of kidney and associated disorders, as well as related molecular genetics, pathophysiology, and epidemiology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


