Tianming Qu, Steven L. Austin, Lianqing Zheng, James Zhang and Wei Yang*,
{"title":"通过分子动力学模拟中的构象动力学捕获和水基口袋表征鉴定蛋白质隐位","authors":"Tianming Qu, Steven L. Austin, Lianqing Zheng, James Zhang and Wei Yang*, ","doi":"10.1021/acs.jctc.5c01019","DOIUrl":null,"url":null,"abstract":"<p >Employing molecular dynamics (MD) simulation to study the formation of novel protein cryptic sites has attracted increasing interest in the field of drug discovery. One specific challenge in this area is finding a viable method to accurately identify and characterize cryptic site transitions from MD simulation results while minimizing the need for extensive human input. Since the formation of cryptic sites often involves significant conformational changes in the protein structure, a method capable of capturing and describing these dynamic pocket transitions with precision is essential. In this paper, we present a new procedure, Conformational Dynamics Capturing and Water-Based Characterization (CDC-WBC). This procedure dynamically identifies the cryptic site region by tracking protein conformational changes observed during molecular dynamics (MD) simulations. The procedure also incorporates water density information to enhance the characterization of cryptic sites across frames. We evaluate the CDC-WBC procedure by applying it to characterize the opening process of the two well-studied cryptic sites in TEM1 β-lactamase. The results demonstrate that the CDC-WBC method accurately captures the open-closed transitions of the two cryptic sites. For comparison, three commonly used protein cavity detection methods in cryptic sites studies, POVME2, Epock, and MDpocket, are applied to identify the “CBT” cryptic site in TEM1 β-lactamase. The results show that the CDC-WBC method outperforms these methods in characterizing the transitions of the “CBT” cryptic site. Additionally, using a benchmark set of 84 protein systems (93 cryptic pockets) from the CryptoSite data set, CDC-WBC consistently shows better performance in distinguishing between the open and closed states of cryptic sites, further highlighting its capability for precise characterization of dynamic cryptic site transitions. The detailed implementation of the CDC-WBC procedure and demo data sets are uploaded to GitHub: https://github.com/TianmingQu/CDC-WBC.git</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"21 17","pages":"8571–8582"},"PeriodicalIF":5.5000,"publicationDate":"2025-08-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Identification of Protein Cryptic Sites via Conformational Dynamics Capturing and Water-Based Pocket Characterization in Molecular Dynamics Simulations\",\"authors\":\"Tianming Qu, Steven L. Austin, Lianqing Zheng, James Zhang and Wei Yang*, \",\"doi\":\"10.1021/acs.jctc.5c01019\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Employing molecular dynamics (MD) simulation to study the formation of novel protein cryptic sites has attracted increasing interest in the field of drug discovery. One specific challenge in this area is finding a viable method to accurately identify and characterize cryptic site transitions from MD simulation results while minimizing the need for extensive human input. Since the formation of cryptic sites often involves significant conformational changes in the protein structure, a method capable of capturing and describing these dynamic pocket transitions with precision is essential. In this paper, we present a new procedure, Conformational Dynamics Capturing and Water-Based Characterization (CDC-WBC). This procedure dynamically identifies the cryptic site region by tracking protein conformational changes observed during molecular dynamics (MD) simulations. The procedure also incorporates water density information to enhance the characterization of cryptic sites across frames. We evaluate the CDC-WBC procedure by applying it to characterize the opening process of the two well-studied cryptic sites in TEM1 β-lactamase. The results demonstrate that the CDC-WBC method accurately captures the open-closed transitions of the two cryptic sites. For comparison, three commonly used protein cavity detection methods in cryptic sites studies, POVME2, Epock, and MDpocket, are applied to identify the “CBT” cryptic site in TEM1 β-lactamase. The results show that the CDC-WBC method outperforms these methods in characterizing the transitions of the “CBT” cryptic site. Additionally, using a benchmark set of 84 protein systems (93 cryptic pockets) from the CryptoSite data set, CDC-WBC consistently shows better performance in distinguishing between the open and closed states of cryptic sites, further highlighting its capability for precise characterization of dynamic cryptic site transitions. The detailed implementation of the CDC-WBC procedure and demo data sets are uploaded to GitHub: https://github.com/TianmingQu/CDC-WBC.git</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"21 17\",\"pages\":\"8571–8582\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2025-08-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jctc.5c01019\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jctc.5c01019","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Identification of Protein Cryptic Sites via Conformational Dynamics Capturing and Water-Based Pocket Characterization in Molecular Dynamics Simulations
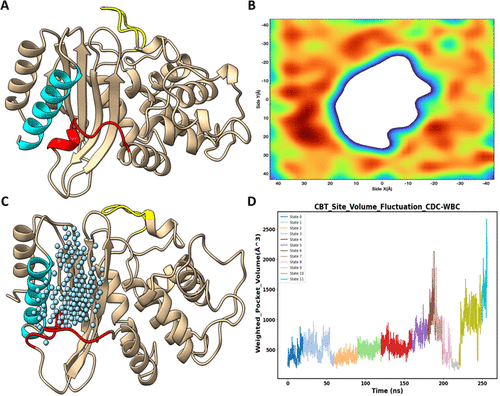
Employing molecular dynamics (MD) simulation to study the formation of novel protein cryptic sites has attracted increasing interest in the field of drug discovery. One specific challenge in this area is finding a viable method to accurately identify and characterize cryptic site transitions from MD simulation results while minimizing the need for extensive human input. Since the formation of cryptic sites often involves significant conformational changes in the protein structure, a method capable of capturing and describing these dynamic pocket transitions with precision is essential. In this paper, we present a new procedure, Conformational Dynamics Capturing and Water-Based Characterization (CDC-WBC). This procedure dynamically identifies the cryptic site region by tracking protein conformational changes observed during molecular dynamics (MD) simulations. The procedure also incorporates water density information to enhance the characterization of cryptic sites across frames. We evaluate the CDC-WBC procedure by applying it to characterize the opening process of the two well-studied cryptic sites in TEM1 β-lactamase. The results demonstrate that the CDC-WBC method accurately captures the open-closed transitions of the two cryptic sites. For comparison, three commonly used protein cavity detection methods in cryptic sites studies, POVME2, Epock, and MDpocket, are applied to identify the “CBT” cryptic site in TEM1 β-lactamase. The results show that the CDC-WBC method outperforms these methods in characterizing the transitions of the “CBT” cryptic site. Additionally, using a benchmark set of 84 protein systems (93 cryptic pockets) from the CryptoSite data set, CDC-WBC consistently shows better performance in distinguishing between the open and closed states of cryptic sites, further highlighting its capability for precise characterization of dynamic cryptic site transitions. The detailed implementation of the CDC-WBC procedure and demo data sets are uploaded to GitHub: https://github.com/TianmingQu/CDC-WBC.git
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


