细胞周期蛋白A/B RxL抑制剂靶向g1 - s检查点受损的癌症
IF 48.5
1区 综合性期刊
Q1 MULTIDISCIPLINARY SCIENCES
引用次数: 0
摘要
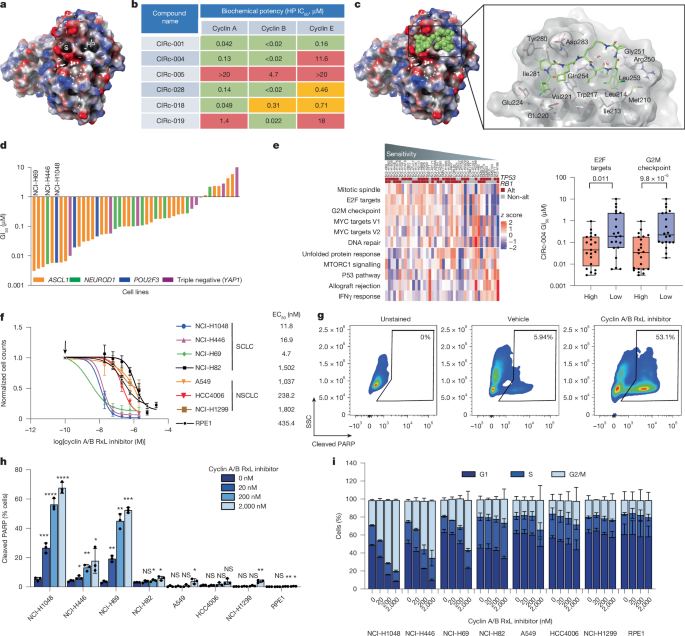
小细胞肺癌(sclc)包含RB1和TP53几乎普遍的功能缺失突变,损害G1-S检查点并导致E2F活性失调1。其他癌症类似地通过CDKN2A的缺失或细胞周期蛋白D或细胞周期蛋白E的扩增破坏G1-S检查点,也导致过度的E2F活性2,3。尽管E2F激活对于细胞周期进程至关重要,但过度激活会促进细胞凋亡4,5,6,7,8,9,呈现出治疗脆弱性。细胞周期蛋白使用保守的疏水性贴片与带有短线性RxL基序的底物结合10,11,12,13。Cyclin A通过依赖rxl的相互作用抑制E2F 10,14,当这种相互作用被破坏时,会过度激活E2F15。然而,这种基板界面仍然难以定位。在这里,我们开发了细胞渗透性,口服生物可利用的大环肽,抑制rxl介导的细胞周期蛋白与其底物的相互作用。cyclin A和cyclin B RxL基序的双重抑制剂(cyclin A/Bi)选择性地杀死SCLC细胞和其他具有高E2F活性的癌细胞。基因筛选显示,细胞周期蛋白A/Bi通过细胞周期蛋白B-和cdk2依赖性纺锤体组装检查点激活诱导细胞凋亡。从机制上讲,cyclin A/Bi通过阻断cyclin A - E2F和cyclin B - myt1 RxL相互作用来过度激活E2F和cyclin B。值得注意的是,周期蛋白A/Bi促进了新形态周期蛋白B-CDK2复合物的形成,从而驱动纺锤体组装检查点激活和有丝分裂细胞死亡。最后,口服周期蛋白A/Bi在化疗耐药的SCLC患者来源的异种移植物中显示出强大的抗肿瘤活性。这些发现揭示了细胞周期蛋白A/Bi触发细胞凋亡的功能获得机制,并支持它们在e2f驱动的癌症中发展。本文章由计算机程序翻译,如有差异,请以英文原文为准。

Targeting G1–S-checkpoint-compromised cancers with cyclin A/B RxL inhibitors
Small-cell lung cancers (SCLCs) contain near-universal loss-of-function mutations in RB1 and TP53, compromising the G1–S checkpoint and leading to dysregulated E2F activity1. Other cancers similarly disrupt the G1–S checkpoint through loss of CDKN2A or amplification of cyclin D or cyclin E, also resulting in excessive E2F activity2,3. Although E2F activation is essential for cell cycle progression, hyperactivation promotes apoptosis4–9, presenting a therapeutic vulnerability. Cyclin proteins use a conserved hydrophobic patch to bind to substrates bearing short linear RxL motifs10–13. Cyclin A represses E2F through an RxL-dependent interaction10,14, which, when disrupted, hyperactivates E2F15. However, this substrate interface has remained difficult to target. Here we developed cell-permeable, orally bioavailable macrocyclic peptides that inhibit RxL-mediated interactions of cyclins with their substrates. Dual inhibitors of cyclin A and cyclin B RxL motifs (cyclin A/Bi) selectively kill SCLC cells and other cancer cells with high E2F activity. Genetic screens revealed that cyclin A/Bi induces apoptosis through cyclin B- and CDK2-dependent spindle assembly checkpoint activation. Mechanistically, cyclin A/Bi hyperactivates E2F and cyclin B by blocking cyclin A–E2F and cyclin B–MYT1 RxL interactions. Notably, cyclin A/Bi promoted the formation of neomorphic cyclin B–CDK2 complexes, which drive spindle assembly checkpoint activation and mitotic cell death. Finally, orally administered cyclin A/Bi showed robust anti-tumour activity in chemotherapy-resistant SCLC patient-derived xenografts. These findings reveal gain-of-function mechanisms through which cyclin A/Bi triggers apoptosis and support their development for E2F-driven cancers. Dual cyclin A/B RxL inhibitors selectively kill small cell lung cancer cells and other cancer cells with high E2F activity.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Nature
综合性期刊-综合性期刊
CiteScore
90.00
自引率
1.20%
发文量
3652
审稿时长
3 months
期刊介绍:
Nature is a prestigious international journal that publishes peer-reviewed research in various scientific and technological fields. The selection of articles is based on criteria such as originality, importance, interdisciplinary relevance, timeliness, accessibility, elegance, and surprising conclusions. In addition to showcasing significant scientific advances, Nature delivers rapid, authoritative, insightful news, and interpretation of current and upcoming trends impacting science, scientists, and the broader public. The journal serves a dual purpose: firstly, to promptly share noteworthy scientific advances and foster discussions among scientists, and secondly, to ensure the swift dissemination of scientific results globally, emphasizing their significance for knowledge, culture, and daily life.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


