Evette B.M. Hillman , Danielle Carson , Julian R.F. Walters , Martin Fritzsche , Ryan Mate , Katie E. Chappell , Elena Chekmeneva , Maria Gomez Romero , Stephen J. Lewis , Sjoerd Rijpkema , Elizabeth M.H. Wellington , Ramesh Arasaradnam , Gregory C.A. Amos
{"title":"原发性胆汁酸性腹泻患者粪便中的瘤状球菌和生物膜标志物提示新的疾病机制和诊断检测潜力","authors":"Evette B.M. Hillman , Danielle Carson , Julian R.F. Walters , Martin Fritzsche , Ryan Mate , Katie E. Chappell , Elena Chekmeneva , Maria Gomez Romero , Stephen J. Lewis , Sjoerd Rijpkema , Elizabeth M.H. Wellington , Ramesh Arasaradnam , Gregory C.A. Amos","doi":"10.1016/j.gastha.2025.100712","DOIUrl":null,"url":null,"abstract":"<div><h3>Background and Aims</h3><div>Bile acid diarrhea (BAD) is a common cause of frequent loose stools, urgency, and incontinence, which is under-recognized due to limited diagnostic test availability and unclear pathogenesis. This study aimed to investigate fecal changes in well-defined subjects.</div></div><div><h3>Methods</h3><div>Fecal samples were compared in BAD patients (n = 26), diagnosed by SeHCAT testing, and healthy controls (n = 21). Shotgun metagenomic sequencing was used to identify microbiome species and functional genes. An extended set of 38 bile acids was quantified by liquid chromatography mass spectrometry, including various epimers and intermediates, such as iso- (3-beta-OH), oxo (keto), allo (5-alpha), and 3-sulfated forms.</div></div><div><h3>Results</h3><div>Alpha diversity, reflecting microbial richness, was reduced in BAD patients with severe forms of the disease, while beta diversity demonstrated distinct microbial profiles between groups. <em>Ruminococcus gnavus</em> (<em>R. gnavus</em>) was prevalent in BAD patients but rare in controls (odds ratio = 73), while <em>Firmicutes bacterium</em> CAG110, <em>Eubacterium siraeum</em> and 2 <em>Oscillibacter</em> species were less common in BAD (odds ratios = 25–30). Overall, 99 taxa differed significantly between groups. Bile acidtransforming genes (<em>baiA</em>, <em>baiB</em>, <em>hdhA</em>) were more abundant in BAD samples (<em>P</em> ≤ .0012). Most fecal bile acids, including iso-bile acids and intermediates, were higher in BAD. Elevated ursodeoxycholic acid-3-sulfate and relatively lower lithocholic acid and allo-bile acids, including isoallolithocholic acid, reflect changes in bacterial metabolism. Biofilm-associated genes (<em>bssS</em>, <em>pgaA</em>, <em>pgaB</em>) were markedly elevated in BAD patients (<em>P</em> ≤ .00008). SeHCAT values negatively correlated with <em>R. gnavus</em> (rho −0.53, <em>P</em> = .008) and positively with <em>E</em><em>ubacterium siraeum</em> (rho 0.41, <em>P</em> = .041).</div></div><div><h3>Conclusion</h3><div>BAD may result from an overgrowth of <em>R. gnavus</em>, associated with intestinal biofilms and an altered bile acid metabolism.</div></div>","PeriodicalId":73130,"journal":{"name":"Gastro hep advances","volume":"4 9","pages":"Article 100712"},"PeriodicalIF":0.0000,"publicationDate":"2025-01-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Ruminococcus gnavus and Biofilm Markers in Feces From Primary Bile Acid Diarrhea Patients Indicate New Disease Mechanisms and Potential for Diagnostic Testing\",\"authors\":\"Evette B.M. Hillman , Danielle Carson , Julian R.F. Walters , Martin Fritzsche , Ryan Mate , Katie E. Chappell , Elena Chekmeneva , Maria Gomez Romero , Stephen J. Lewis , Sjoerd Rijpkema , Elizabeth M.H. Wellington , Ramesh Arasaradnam , Gregory C.A. Amos\",\"doi\":\"10.1016/j.gastha.2025.100712\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Background and Aims</h3><div>Bile acid diarrhea (BAD) is a common cause of frequent loose stools, urgency, and incontinence, which is under-recognized due to limited diagnostic test availability and unclear pathogenesis. This study aimed to investigate fecal changes in well-defined subjects.</div></div><div><h3>Methods</h3><div>Fecal samples were compared in BAD patients (n = 26), diagnosed by SeHCAT testing, and healthy controls (n = 21). Shotgun metagenomic sequencing was used to identify microbiome species and functional genes. An extended set of 38 bile acids was quantified by liquid chromatography mass spectrometry, including various epimers and intermediates, such as iso- (3-beta-OH), oxo (keto), allo (5-alpha), and 3-sulfated forms.</div></div><div><h3>Results</h3><div>Alpha diversity, reflecting microbial richness, was reduced in BAD patients with severe forms of the disease, while beta diversity demonstrated distinct microbial profiles between groups. <em>Ruminococcus gnavus</em> (<em>R. gnavus</em>) was prevalent in BAD patients but rare in controls (odds ratio = 73), while <em>Firmicutes bacterium</em> CAG110, <em>Eubacterium siraeum</em> and 2 <em>Oscillibacter</em> species were less common in BAD (odds ratios = 25–30). Overall, 99 taxa differed significantly between groups. Bile acidtransforming genes (<em>baiA</em>, <em>baiB</em>, <em>hdhA</em>) were more abundant in BAD samples (<em>P</em> ≤ .0012). Most fecal bile acids, including iso-bile acids and intermediates, were higher in BAD. Elevated ursodeoxycholic acid-3-sulfate and relatively lower lithocholic acid and allo-bile acids, including isoallolithocholic acid, reflect changes in bacterial metabolism. Biofilm-associated genes (<em>bssS</em>, <em>pgaA</em>, <em>pgaB</em>) were markedly elevated in BAD patients (<em>P</em> ≤ .00008). SeHCAT values negatively correlated with <em>R. gnavus</em> (rho −0.53, <em>P</em> = .008) and positively with <em>E</em><em>ubacterium siraeum</em> (rho 0.41, <em>P</em> = .041).</div></div><div><h3>Conclusion</h3><div>BAD may result from an overgrowth of <em>R. gnavus</em>, associated with intestinal biofilms and an altered bile acid metabolism.</div></div>\",\"PeriodicalId\":73130,\"journal\":{\"name\":\"Gastro hep advances\",\"volume\":\"4 9\",\"pages\":\"Article 100712\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2025-01-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Gastro hep advances\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2772572325000998\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Gastro hep advances","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2772572325000998","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
摘要
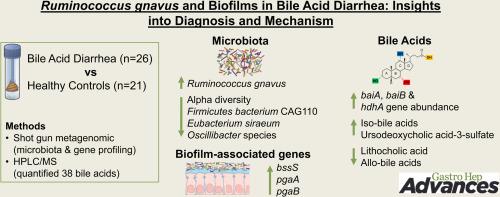
背景和目的胆汁酸性腹泻(BAD)是一种常见的引起频繁稀便、尿急和尿失禁的原因,由于诊断测试的有限性和发病机制不明确,对其认识不足。本研究旨在调查明确受试者的粪便变化。方法将经SeHCAT检测诊断的BAD患者(26例)与健康对照组(21例)的粪便标本进行比较。采用散弹枪宏基因组测序技术鉴定微生物组种类和功能基因。采用液相色谱质谱法对38种胆汁酸进行了定量分析,包括各种外显体和中间体,如异- (3- β - oh)、氧(酮)、允许(5- α)和3-硫酸酸化形式。结果反映微生物丰富度的β多样性在严重形式的BAD患者中减少,而β多样性在组间表现出不同的微生物特征。瘤胃球菌(R. gnavus)在BAD患者中普遍存在,但在对照组中罕见(优势比= 73),而厚壁菌门细菌CAG110、西拉真杆菌和2种Oscillibacter在BAD中较少见(优势比= 25-30)。总体而言,99个类群在组间存在显著差异。胆汁酸转化基因(baiA、baiB、hdhA)在BAD样品中含量较高(P≤0.0012)。大多数粪便胆汁酸,包括异胆汁酸和中间产物,在BAD中含量较高。升高的熊去氧胆酸-3-硫酸盐和相对较低的石胆酸和异丙胆酸,包括异丙胆酸,反映了细菌代谢的变化。BAD患者生物膜相关基因(bssS、pgaA、pgaB)明显升高(P≤0.008)。SeHCAT值与gnavus呈负相关(rho - 0.53, P = 0.008),与siraeum呈正相关(rho 0.41, P = 0.041)。结论bad可能是由鼠的过度生长引起的,与肠道生物膜和胆汁酸代谢改变有关。
Ruminococcus gnavus and Biofilm Markers in Feces From Primary Bile Acid Diarrhea Patients Indicate New Disease Mechanisms and Potential for Diagnostic Testing
Background and Aims
Bile acid diarrhea (BAD) is a common cause of frequent loose stools, urgency, and incontinence, which is under-recognized due to limited diagnostic test availability and unclear pathogenesis. This study aimed to investigate fecal changes in well-defined subjects.
Methods
Fecal samples were compared in BAD patients (n = 26), diagnosed by SeHCAT testing, and healthy controls (n = 21). Shotgun metagenomic sequencing was used to identify microbiome species and functional genes. An extended set of 38 bile acids was quantified by liquid chromatography mass spectrometry, including various epimers and intermediates, such as iso- (3-beta-OH), oxo (keto), allo (5-alpha), and 3-sulfated forms.
Results
Alpha diversity, reflecting microbial richness, was reduced in BAD patients with severe forms of the disease, while beta diversity demonstrated distinct microbial profiles between groups. Ruminococcus gnavus (R. gnavus) was prevalent in BAD patients but rare in controls (odds ratio = 73), while Firmicutes bacterium CAG110, Eubacterium siraeum and 2 Oscillibacter species were less common in BAD (odds ratios = 25–30). Overall, 99 taxa differed significantly between groups. Bile acidtransforming genes (baiA, baiB, hdhA) were more abundant in BAD samples (P ≤ .0012). Most fecal bile acids, including iso-bile acids and intermediates, were higher in BAD. Elevated ursodeoxycholic acid-3-sulfate and relatively lower lithocholic acid and allo-bile acids, including isoallolithocholic acid, reflect changes in bacterial metabolism. Biofilm-associated genes (bssS, pgaA, pgaB) were markedly elevated in BAD patients (P ≤ .00008). SeHCAT values negatively correlated with R. gnavus (rho −0.53, P = .008) and positively with Eubacterium siraeum (rho 0.41, P = .041).
Conclusion
BAD may result from an overgrowth of R. gnavus, associated with intestinal biofilms and an altered bile acid metabolism.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


