{"title":"中国线粒体神经胃肠道脑肌病:一种新的TYMP变异和全面的临床遗传学见解。","authors":"Xuebi Xu, Junhui Xia, Fei Xu, Mingshan Wang, Lihong Yang, Xiaoli Chen","doi":"10.1186/s13023-025-03962-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Mitochondrial neurogastrointestinal encephalopathy (MNGIE) is a rare autosomal recessive disorder caused by variants in the TYMP gene, which encodes thymidine phosphorylase (TP). It is characterized by multisystem involvement, with prominent gastrointestinal, neurological, and systemic manifestations that typically exhibit progressive worsening over time.</p><p><strong>Methods: </strong>We characterized a multigenerational MNGIE family through comprehensive proband analysis, identifying compound heterozygous TYMP variants (c.131G > C, p.Arg44Pro and c.1268T>G, p.Leu423Arg in trans) as the molecular basis of disease. Extended family testing for genetic counseling confirmed no secondary pathogenic variants. Muscle biopsies were analyzed using comprehensive staining techniques. Genomic analysis involved next-generation sequencing (NGS) of the proband's DNA and Sanger sequencing of family members' DNA to confirm variants. In silico analysis utilized bioinformatics tools and protein modeling to predict pathogenicity and assess structural impacts, with variant classification adhering to American College of Medical Genetics and Genomics(ACMG) guidelines. Additionally, a literature review of Chinese MNGIE cases was conducted to contextualize the findings.</p><p><strong>Results: </strong>The proband exhibited characteristic MNGIE features, including gastrointestinal dysmotility, diffuse leukoencephalopathy on brain MRI (magnetic resonance imaging), and electrophysiologically confirmed peripheral neuropathy. Muscle biopsy revealed ragged red fibers, cytochrome c oxidase-deficient fibers, and enhanced succinate dehydrogenase activity in blood vessels, consistent with mitochondrial dysfunction. Genetic analysis identified a novel TYMP variant (c.1268T > G, p.Leu423Arg) and a known variant (c.131G > C, p.Arg44Pro) in the proband, both classified as likely pathogenic according to ACMG guidelines. Molecular analysis of other 11 family members detected heterozygous carriers of either the c.1268T > G or c.131G > C variant in six asymptomatic individuals. In silico analysis confirmed that both variants are highly conserved and likely pathogenic. Protein modeling revealed that both variants compromise structural integrity and conformation, impairing TP function. Homozygous or compound heterozygous missense variants were identified as the predominant genetic alterations in 16 Chinese MNGIE cases, with gastrointestinal and neurological symptoms being the most common clinical manifestations.</p><p><strong>Conclusions: </strong>This study enriches the variant spectrum in Chinese patients, highlights the importance of early diagnosis prior to the onset of cachexia and irreversible tissue damage, and enhances the understanding of genetic heterogeneity.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"439"},"PeriodicalIF":3.5000,"publicationDate":"2025-08-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12363080/pdf/","citationCount":"0","resultStr":"{\"title\":\"Mitochondrial neurogastrointestinal encephalomyopathy in china: a novel TYMP variant and comprehensive clinical-genetic insights.\",\"authors\":\"Xuebi Xu, Junhui Xia, Fei Xu, Mingshan Wang, Lihong Yang, Xiaoli Chen\",\"doi\":\"10.1186/s13023-025-03962-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Mitochondrial neurogastrointestinal encephalopathy (MNGIE) is a rare autosomal recessive disorder caused by variants in the TYMP gene, which encodes thymidine phosphorylase (TP). It is characterized by multisystem involvement, with prominent gastrointestinal, neurological, and systemic manifestations that typically exhibit progressive worsening over time.</p><p><strong>Methods: </strong>We characterized a multigenerational MNGIE family through comprehensive proband analysis, identifying compound heterozygous TYMP variants (c.131G > C, p.Arg44Pro and c.1268T>G, p.Leu423Arg in trans) as the molecular basis of disease. Extended family testing for genetic counseling confirmed no secondary pathogenic variants. Muscle biopsies were analyzed using comprehensive staining techniques. Genomic analysis involved next-generation sequencing (NGS) of the proband's DNA and Sanger sequencing of family members' DNA to confirm variants. In silico analysis utilized bioinformatics tools and protein modeling to predict pathogenicity and assess structural impacts, with variant classification adhering to American College of Medical Genetics and Genomics(ACMG) guidelines. Additionally, a literature review of Chinese MNGIE cases was conducted to contextualize the findings.</p><p><strong>Results: </strong>The proband exhibited characteristic MNGIE features, including gastrointestinal dysmotility, diffuse leukoencephalopathy on brain MRI (magnetic resonance imaging), and electrophysiologically confirmed peripheral neuropathy. Muscle biopsy revealed ragged red fibers, cytochrome c oxidase-deficient fibers, and enhanced succinate dehydrogenase activity in blood vessels, consistent with mitochondrial dysfunction. Genetic analysis identified a novel TYMP variant (c.1268T > G, p.Leu423Arg) and a known variant (c.131G > C, p.Arg44Pro) in the proband, both classified as likely pathogenic according to ACMG guidelines. Molecular analysis of other 11 family members detected heterozygous carriers of either the c.1268T > G or c.131G > C variant in six asymptomatic individuals. In silico analysis confirmed that both variants are highly conserved and likely pathogenic. Protein modeling revealed that both variants compromise structural integrity and conformation, impairing TP function. Homozygous or compound heterozygous missense variants were identified as the predominant genetic alterations in 16 Chinese MNGIE cases, with gastrointestinal and neurological symptoms being the most common clinical manifestations.</p><p><strong>Conclusions: </strong>This study enriches the variant spectrum in Chinese patients, highlights the importance of early diagnosis prior to the onset of cachexia and irreversible tissue damage, and enhances the understanding of genetic heterogeneity.</p>\",\"PeriodicalId\":19651,\"journal\":{\"name\":\"Orphanet Journal of Rare Diseases\",\"volume\":\"20 1\",\"pages\":\"439\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2025-08-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12363080/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Orphanet Journal of Rare Diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13023-025-03962-3\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-03962-3","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Mitochondrial neurogastrointestinal encephalomyopathy in china: a novel TYMP variant and comprehensive clinical-genetic insights.
Background: Mitochondrial neurogastrointestinal encephalopathy (MNGIE) is a rare autosomal recessive disorder caused by variants in the TYMP gene, which encodes thymidine phosphorylase (TP). It is characterized by multisystem involvement, with prominent gastrointestinal, neurological, and systemic manifestations that typically exhibit progressive worsening over time.
Methods: We characterized a multigenerational MNGIE family through comprehensive proband analysis, identifying compound heterozygous TYMP variants (c.131G > C, p.Arg44Pro and c.1268T>G, p.Leu423Arg in trans) as the molecular basis of disease. Extended family testing for genetic counseling confirmed no secondary pathogenic variants. Muscle biopsies were analyzed using comprehensive staining techniques. Genomic analysis involved next-generation sequencing (NGS) of the proband's DNA and Sanger sequencing of family members' DNA to confirm variants. In silico analysis utilized bioinformatics tools and protein modeling to predict pathogenicity and assess structural impacts, with variant classification adhering to American College of Medical Genetics and Genomics(ACMG) guidelines. Additionally, a literature review of Chinese MNGIE cases was conducted to contextualize the findings.
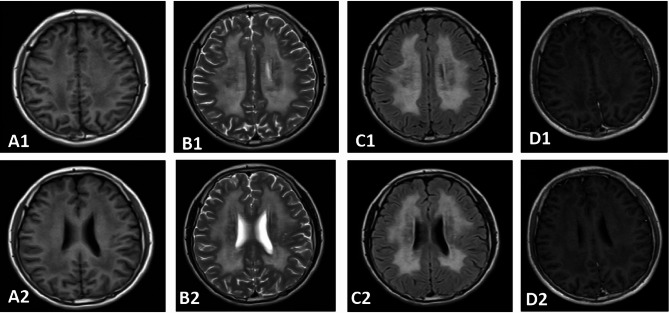
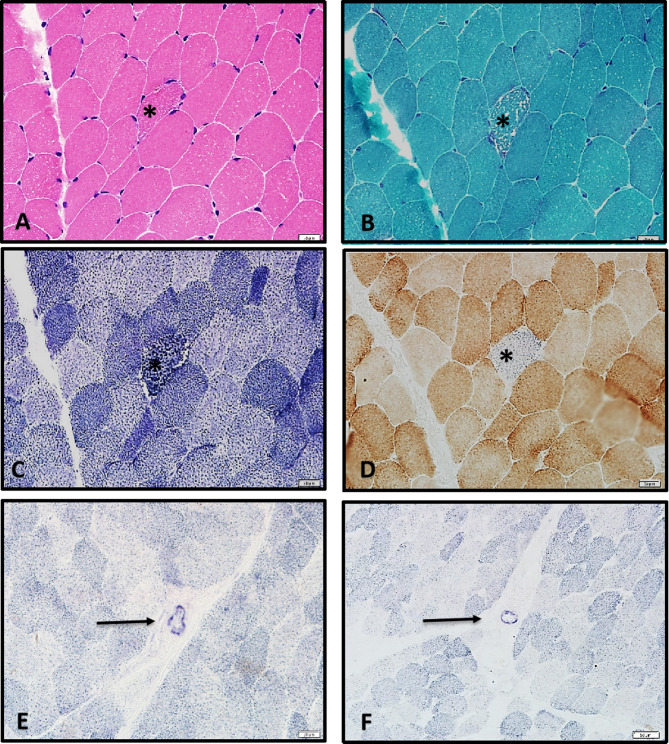
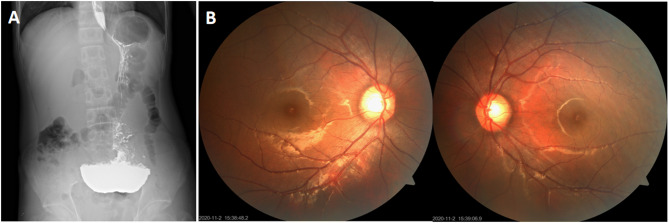
Results: The proband exhibited characteristic MNGIE features, including gastrointestinal dysmotility, diffuse leukoencephalopathy on brain MRI (magnetic resonance imaging), and electrophysiologically confirmed peripheral neuropathy. Muscle biopsy revealed ragged red fibers, cytochrome c oxidase-deficient fibers, and enhanced succinate dehydrogenase activity in blood vessels, consistent with mitochondrial dysfunction. Genetic analysis identified a novel TYMP variant (c.1268T > G, p.Leu423Arg) and a known variant (c.131G > C, p.Arg44Pro) in the proband, both classified as likely pathogenic according to ACMG guidelines. Molecular analysis of other 11 family members detected heterozygous carriers of either the c.1268T > G or c.131G > C variant in six asymptomatic individuals. In silico analysis confirmed that both variants are highly conserved and likely pathogenic. Protein modeling revealed that both variants compromise structural integrity and conformation, impairing TP function. Homozygous or compound heterozygous missense variants were identified as the predominant genetic alterations in 16 Chinese MNGIE cases, with gastrointestinal and neurological symptoms being the most common clinical manifestations.
Conclusions: This study enriches the variant spectrum in Chinese patients, highlights the importance of early diagnosis prior to the onset of cachexia and irreversible tissue damage, and enhances the understanding of genetic heterogeneity.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


