{"title":"在中国GNAO1脑病队列中鉴定三种新的GNAO1变异:扩大临床和遗传谱","authors":"Daoqi Mei, Yu Gu, Bingbing Zhang, Shiyue Mei, Xiaona Wang, Yuanning Ma, Jie Deng, Jihong Tang","doi":"10.1186/s13023-025-03984-x","DOIUrl":null,"url":null,"abstract":"<p><strong>Objective: </strong>To summarize the clinical characteristics of a cohort of nine Chinese children with GNAO1 encephalopathy and analyze their genotypes.</p><p><strong>Methods: </strong>A retrospective study was conducted on nine children diagnosed with GNAO1 encephalopathy at the Neurology Department of two children's hospitals between January 2019 and December 2022. Their clinical manifestations, genetic test results, cranial imaging, electroencephalography and treatment were summarized. Their prognosis was followed up.</p><p><strong>Results: </strong>All nine patients presented with moderate-to-severe psychomotor developmental delay and dystonia. Six patients exhibited neonatal or infantile-onset epilepsy, manifesting as generalized tonic-clonic seizure, myoclonic seizure, epileptic spasms, and were diagnosed with developmental and epileptic encephalopathy 17 (DEE 17). Two patients presented with choreoathetosis in infancy without epileptic seizure and were diagnosed with the neurodevelopmental disorder with involuntary movements (NEDIM). One patient presented with choreoathetosis at two years of age and developed focal seizures at six years of age, representing an intermediate phenotype. During a follow-up period of 0.8-3.5 years, one child died due to infection. The remaining eight continued to exhibit psychomotor retardation. Pathogenic or likely pathogenic de novo heterozygous missense variants in GNAO1 were identified in all nine cases. Among these, the variants c.17G > T (p.Ser6Ile), c.119G > C (p.Gly40Ala), and c.748 C > T (p.Leu250Phe) are novel.</p><p><strong>Conclusion: </strong>In conclusion, we analyzed the clinical characteristics and genetic variants of a cohort of nine Chinese children with GNAO1 variants and identified three novel GNAO1 variants. Our study expanded the spectrum of genotypes and phenotypes in GNOA1-associated encephalopathy.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"440"},"PeriodicalIF":3.5000,"publicationDate":"2025-08-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12363038/pdf/","citationCount":"0","resultStr":"{\"title\":\"Identification of three novel GNAO1 variants in a Chinese cohort with GNAO1 encephalopathy: expanding the clinical and genetic spectrum.\",\"authors\":\"Daoqi Mei, Yu Gu, Bingbing Zhang, Shiyue Mei, Xiaona Wang, Yuanning Ma, Jie Deng, Jihong Tang\",\"doi\":\"10.1186/s13023-025-03984-x\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Objective: </strong>To summarize the clinical characteristics of a cohort of nine Chinese children with GNAO1 encephalopathy and analyze their genotypes.</p><p><strong>Methods: </strong>A retrospective study was conducted on nine children diagnosed with GNAO1 encephalopathy at the Neurology Department of two children's hospitals between January 2019 and December 2022. Their clinical manifestations, genetic test results, cranial imaging, electroencephalography and treatment were summarized. Their prognosis was followed up.</p><p><strong>Results: </strong>All nine patients presented with moderate-to-severe psychomotor developmental delay and dystonia. Six patients exhibited neonatal or infantile-onset epilepsy, manifesting as generalized tonic-clonic seizure, myoclonic seizure, epileptic spasms, and were diagnosed with developmental and epileptic encephalopathy 17 (DEE 17). Two patients presented with choreoathetosis in infancy without epileptic seizure and were diagnosed with the neurodevelopmental disorder with involuntary movements (NEDIM). One patient presented with choreoathetosis at two years of age and developed focal seizures at six years of age, representing an intermediate phenotype. During a follow-up period of 0.8-3.5 years, one child died due to infection. The remaining eight continued to exhibit psychomotor retardation. Pathogenic or likely pathogenic de novo heterozygous missense variants in GNAO1 were identified in all nine cases. Among these, the variants c.17G > T (p.Ser6Ile), c.119G > C (p.Gly40Ala), and c.748 C > T (p.Leu250Phe) are novel.</p><p><strong>Conclusion: </strong>In conclusion, we analyzed the clinical characteristics and genetic variants of a cohort of nine Chinese children with GNAO1 variants and identified three novel GNAO1 variants. Our study expanded the spectrum of genotypes and phenotypes in GNOA1-associated encephalopathy.</p>\",\"PeriodicalId\":19651,\"journal\":{\"name\":\"Orphanet Journal of Rare Diseases\",\"volume\":\"20 1\",\"pages\":\"440\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2025-08-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12363038/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Orphanet Journal of Rare Diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13023-025-03984-x\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-03984-x","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
目的:总结9例中国GNAO1型脑病患儿的临床特点并分析其基因型。方法:对2019年1月至2022年12月在两家儿童医院神经科诊断为GNAO1脑病的9名儿童进行回顾性研究。对其临床表现、基因检测结果、颅脑影像学、脑电图及治疗进行综述。随访预后。结果:9例患者均出现中重度精神运动发育迟缓和肌张力障碍。6例患者表现为新生儿或婴儿癫痫,表现为全身性强直阵挛性发作、肌阵挛性发作、癫痫性痉挛,诊断为发育性和癫痫性脑病17 (DEE 17)。2例患者在婴儿期无癫痫发作表现为舞蹈病,被诊断为伴有不自主运动的神经发育障碍(NEDIM)。1例患者2岁时出现舞蹈病,6岁时出现局灶性癫痫发作,为中间表型。在0.8-3.5年的随访期间,有1名儿童死于感染。其余8人继续表现出精神运动迟缓。在所有9例病例中均鉴定出GNAO1致病性或可能致病性的新生杂合错义变异。其中,C . 17g > T (p.Ser6Ile)、C . 119g >c (p.Gly40Ala)和C .748C b> T (p.Leu250Phe)是新颖的。结论:本研究分析了9例中国儿童GNAO1变异的临床特征和遗传变异,并鉴定出3种新的GNAO1变异。我们的研究扩大了gnoa1相关脑病的基因型和表型谱。
Identification of three novel GNAO1 variants in a Chinese cohort with GNAO1 encephalopathy: expanding the clinical and genetic spectrum.
Objective: To summarize the clinical characteristics of a cohort of nine Chinese children with GNAO1 encephalopathy and analyze their genotypes.
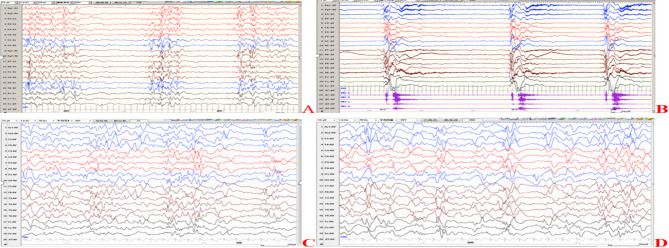
Methods: A retrospective study was conducted on nine children diagnosed with GNAO1 encephalopathy at the Neurology Department of two children's hospitals between January 2019 and December 2022. Their clinical manifestations, genetic test results, cranial imaging, electroencephalography and treatment were summarized. Their prognosis was followed up.
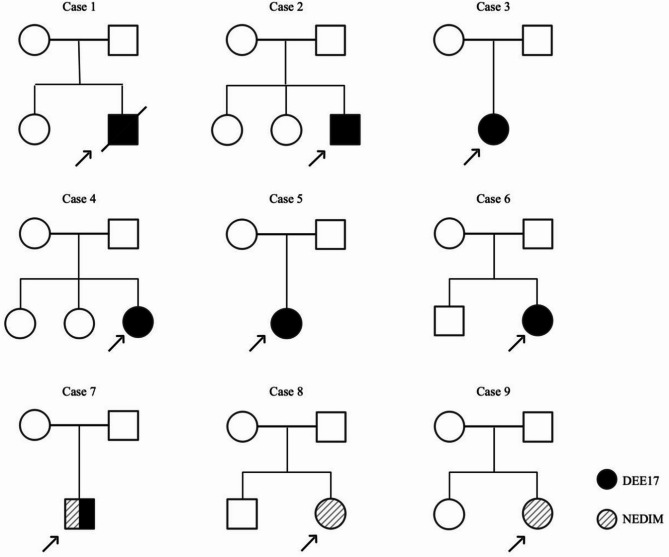
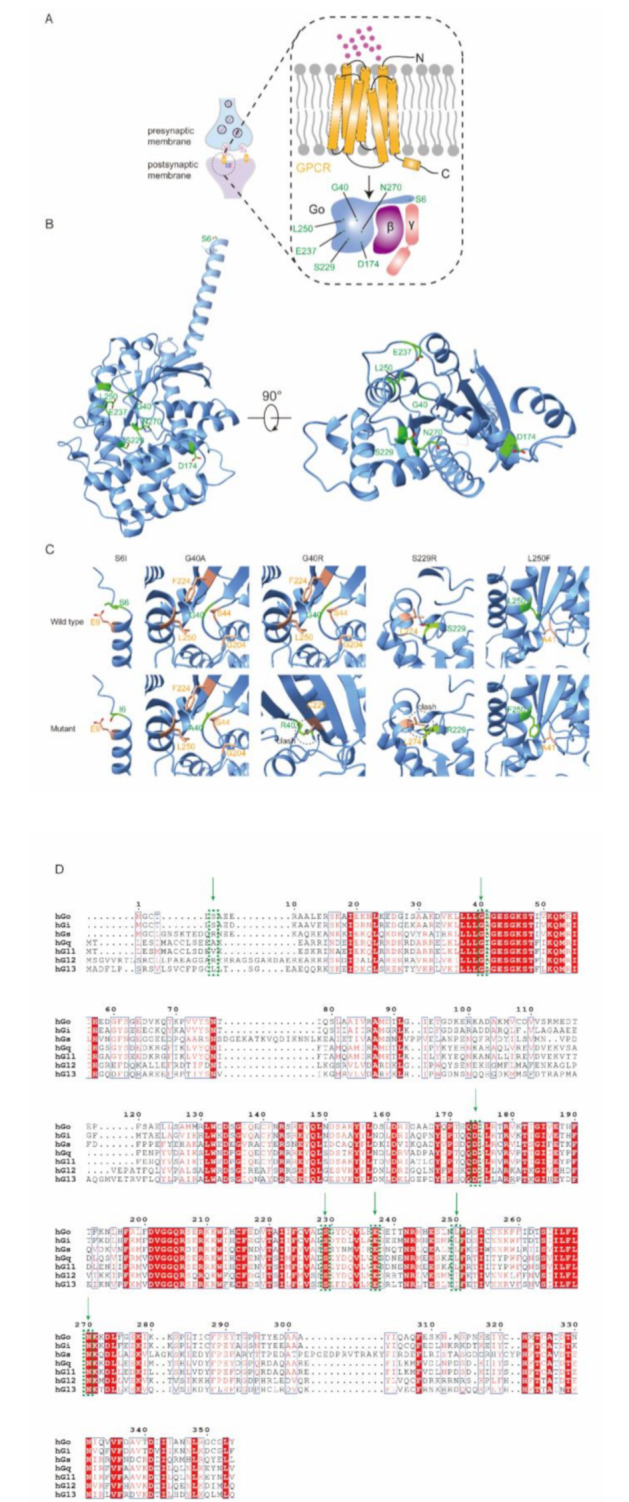
Results: All nine patients presented with moderate-to-severe psychomotor developmental delay and dystonia. Six patients exhibited neonatal or infantile-onset epilepsy, manifesting as generalized tonic-clonic seizure, myoclonic seizure, epileptic spasms, and were diagnosed with developmental and epileptic encephalopathy 17 (DEE 17). Two patients presented with choreoathetosis in infancy without epileptic seizure and were diagnosed with the neurodevelopmental disorder with involuntary movements (NEDIM). One patient presented with choreoathetosis at two years of age and developed focal seizures at six years of age, representing an intermediate phenotype. During a follow-up period of 0.8-3.5 years, one child died due to infection. The remaining eight continued to exhibit psychomotor retardation. Pathogenic or likely pathogenic de novo heterozygous missense variants in GNAO1 were identified in all nine cases. Among these, the variants c.17G > T (p.Ser6Ile), c.119G > C (p.Gly40Ala), and c.748 C > T (p.Leu250Phe) are novel.
Conclusion: In conclusion, we analyzed the clinical characteristics and genetic variants of a cohort of nine Chinese children with GNAO1 variants and identified three novel GNAO1 variants. Our study expanded the spectrum of genotypes and phenotypes in GNOA1-associated encephalopathy.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


