Cyril Jones Jagaraj, Prachi Mehta, Julie Hunter, Julie D Atkin
{"title":"进展较慢的ALS TDP-43 rNLS8小鼠模型:临床前和机制研究的意义","authors":"Cyril Jones Jagaraj, Prachi Mehta, Julie Hunter, Julie D Atkin","doi":"10.1007/s12017-025-08871-z","DOIUrl":null,"url":null,"abstract":"<p><p>Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterised by motor neuron degeneration, muscle weakness, paralysis, and eventual death, with TAR DNA-binding protein 43 (TDP-43) pathology observed in almost all cases. Mouse models based on TDP-43 are thus essential for studying ALS and developing therapeutic approaches. The TDP-43 rNLS8 mouse model expresses a human TDP-43 transgene with a mutated nuclear localization sequence (hTDP-43 ΔNLS), but this is normally suppressed by the presence of doxycycline (Dox). Disease is initiated by removal of Dox, which replicates key ALS features, including TDP-43 pathology, neuromuscular junction denervation, motor neuron loss, and reduced survival. However, this model has a rapid disease progression which limits its use for extended preclinical studies and investigation of early disease mechanisms. To overcome these limitations, we explored whether maintaining low Dox concentrations in the diet (10-20 mg/kg) could slow disease progression. Our findings demonstrate that this approach significantly reduced hTDP-43 ΔNLS expression (up to 4.8-fold), which delayed disease onset by four weeks. Disease progression, assessed by rotarod performance, grip strength, and neurological scores, was extended from six to 15 weeks, with a threefold increase in survival. Despite slower progression, at the end stage, mice displayed similar levels of neuroinflammation, motor neuron loss, as Dox off mice. These findings highlight slower-progressing TDP-43 rNLS8 mice as a robust model for preclinical and early disease mechanism studies.</p>","PeriodicalId":19304,"journal":{"name":"NeuroMolecular Medicine","volume":"27 1","pages":"59"},"PeriodicalIF":3.9000,"publicationDate":"2025-08-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12361281/pdf/","citationCount":"0","resultStr":"{\"title\":\"A Slower-Progressing TDP-43 rNLS8 Mouse Model for ALS: Implications for Preclinical and Mechanistic Studies.\",\"authors\":\"Cyril Jones Jagaraj, Prachi Mehta, Julie Hunter, Julie D Atkin\",\"doi\":\"10.1007/s12017-025-08871-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterised by motor neuron degeneration, muscle weakness, paralysis, and eventual death, with TAR DNA-binding protein 43 (TDP-43) pathology observed in almost all cases. Mouse models based on TDP-43 are thus essential for studying ALS and developing therapeutic approaches. The TDP-43 rNLS8 mouse model expresses a human TDP-43 transgene with a mutated nuclear localization sequence (hTDP-43 ΔNLS), but this is normally suppressed by the presence of doxycycline (Dox). Disease is initiated by removal of Dox, which replicates key ALS features, including TDP-43 pathology, neuromuscular junction denervation, motor neuron loss, and reduced survival. However, this model has a rapid disease progression which limits its use for extended preclinical studies and investigation of early disease mechanisms. To overcome these limitations, we explored whether maintaining low Dox concentrations in the diet (10-20 mg/kg) could slow disease progression. Our findings demonstrate that this approach significantly reduced hTDP-43 ΔNLS expression (up to 4.8-fold), which delayed disease onset by four weeks. Disease progression, assessed by rotarod performance, grip strength, and neurological scores, was extended from six to 15 weeks, with a threefold increase in survival. Despite slower progression, at the end stage, mice displayed similar levels of neuroinflammation, motor neuron loss, as Dox off mice. These findings highlight slower-progressing TDP-43 rNLS8 mice as a robust model for preclinical and early disease mechanism studies.</p>\",\"PeriodicalId\":19304,\"journal\":{\"name\":\"NeuroMolecular Medicine\",\"volume\":\"27 1\",\"pages\":\"59\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2025-08-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12361281/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NeuroMolecular Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1007/s12017-025-08871-z\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"NEUROSCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NeuroMolecular Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s12017-025-08871-z","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
A Slower-Progressing TDP-43 rNLS8 Mouse Model for ALS: Implications for Preclinical and Mechanistic Studies.
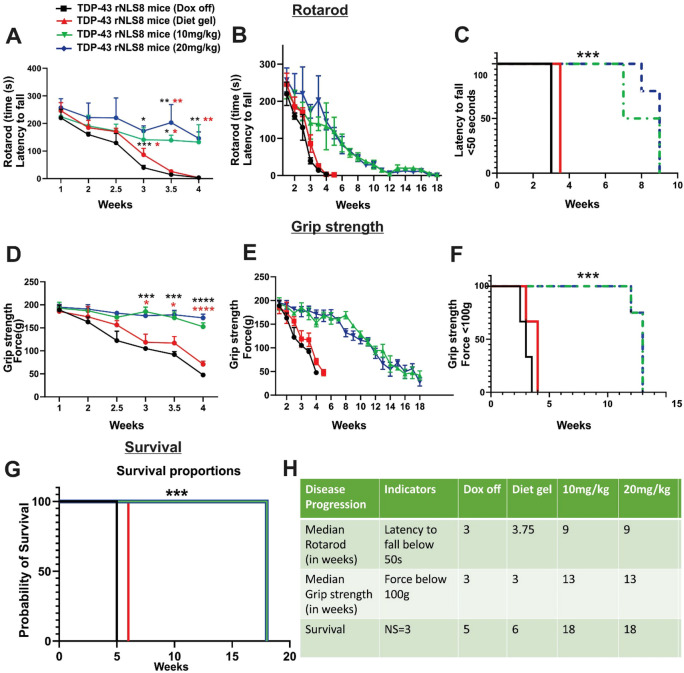
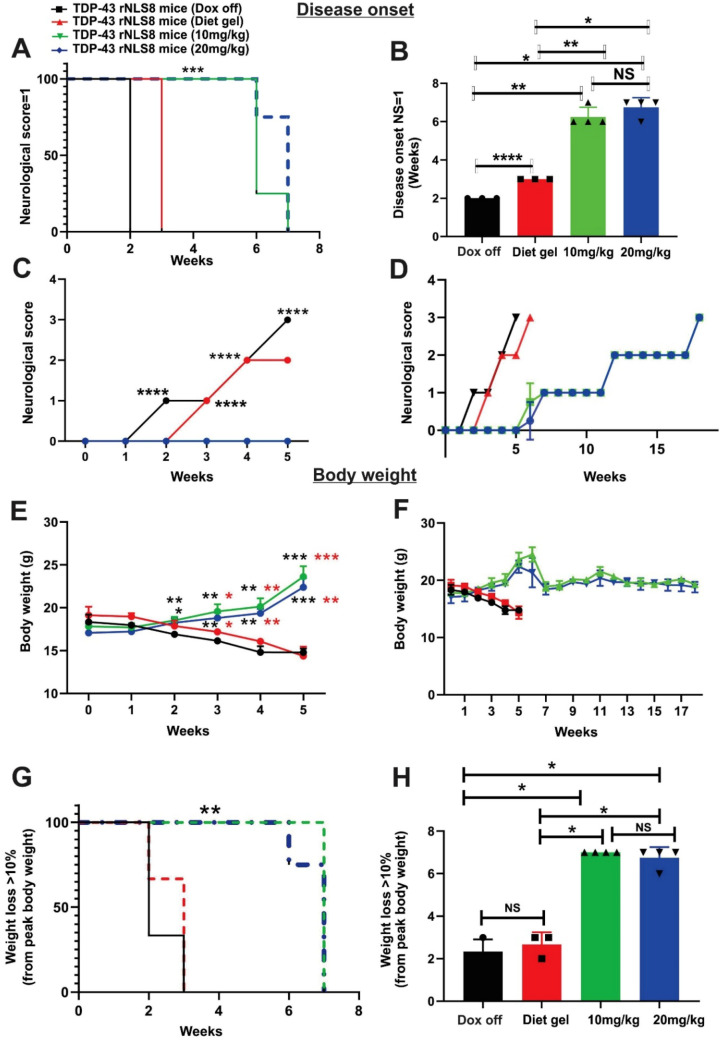
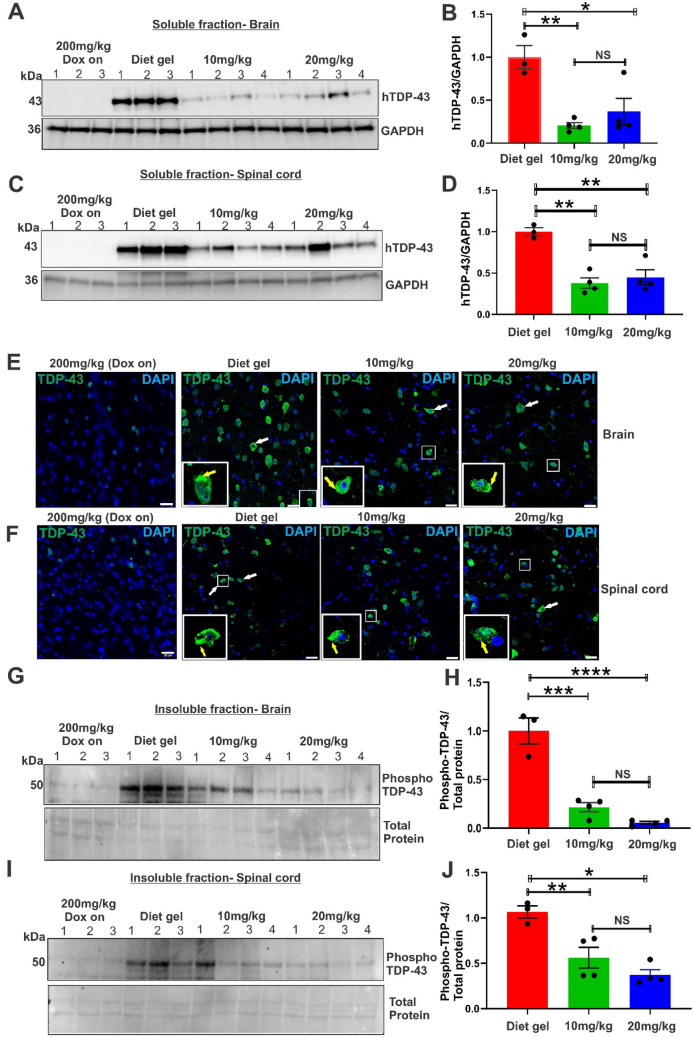
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterised by motor neuron degeneration, muscle weakness, paralysis, and eventual death, with TAR DNA-binding protein 43 (TDP-43) pathology observed in almost all cases. Mouse models based on TDP-43 are thus essential for studying ALS and developing therapeutic approaches. The TDP-43 rNLS8 mouse model expresses a human TDP-43 transgene with a mutated nuclear localization sequence (hTDP-43 ΔNLS), but this is normally suppressed by the presence of doxycycline (Dox). Disease is initiated by removal of Dox, which replicates key ALS features, including TDP-43 pathology, neuromuscular junction denervation, motor neuron loss, and reduced survival. However, this model has a rapid disease progression which limits its use for extended preclinical studies and investigation of early disease mechanisms. To overcome these limitations, we explored whether maintaining low Dox concentrations in the diet (10-20 mg/kg) could slow disease progression. Our findings demonstrate that this approach significantly reduced hTDP-43 ΔNLS expression (up to 4.8-fold), which delayed disease onset by four weeks. Disease progression, assessed by rotarod performance, grip strength, and neurological scores, was extended from six to 15 weeks, with a threefold increase in survival. Despite slower progression, at the end stage, mice displayed similar levels of neuroinflammation, motor neuron loss, as Dox off mice. These findings highlight slower-progressing TDP-43 rNLS8 mice as a robust model for preclinical and early disease mechanism studies.
期刊介绍:
NeuroMolecular Medicine publishes cutting-edge original research articles and critical reviews on the molecular and biochemical basis of neurological disorders. Studies range from genetic analyses of human populations to animal and cell culture models of neurological disorders. Emerging findings concerning the identification of genetic aberrancies and their pathogenic mechanisms at the molecular and cellular levels will be included. Also covered are experimental analyses of molecular cascades involved in the development and adult plasticity of the nervous system, in neurological dysfunction, and in neuronal degeneration and repair. NeuroMolecular Medicine encompasses basic research in the fields of molecular genetics, signal transduction, plasticity, and cell death. The information published in NEMM will provide a window into the future of molecular medicine for the nervous system.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


