Néstor Gutiérrez-Sánchez, Fernando Mendizábal, Sebastián Miranda-Rojas
{"title":"利用量子力学/分子力学和激活-应变模型揭示小氯酶反应机理和选择性。","authors":"Néstor Gutiérrez-Sánchez, Fernando Mendizábal, Sebastián Miranda-Rojas","doi":"10.1002/cplu.202500344","DOIUrl":null,"url":null,"abstract":"<p>Here, an exhaustive exploration of the reaction mechanism toward the chlorination process carried out by SalL, a chlorinase enzyme that catalyzes the conversion of SAM into 5′-chloro-5′-deoxyadenosine through an <i>S</i><sub><i>N</i></sub>2 reaction, is presented. To this end, molecular dynamics simulations and quantum mechanical/molecular mechanics calculations are performed, and 14 density functionals are benchmarked. Among the tested functionals, TPSSh(BJ) provides the closest energy barrier to experimental value. Three configurations of interaction between chloride and the halogen pocket are found, where the best model exhibits a barrier height of 20.1 kcal mol<sup>−1</sup>, close to the 19.9 kcal mol<sup>−1</sup> experimentally obtained. This model is characterized by the chloride interacting with the backbone-amide of Gly131 and Tyr130. The reaction pathway is calculated through the intrinsic reaction coordinate approach, and it is characterized using reaction force analysis and the activation-strain model with energy decomposition analysis to obtain chemical insights into the inner working of this enzyme. According to the main findings, the overstabilization of the halogen binding on the active site increases the barrier height, explaining the lack of activity against fluoride, while the interaction energy between nucleophile−electrophile is responsible of reducing the barrier height, with the orbital interaction energy as the main stabilizing factor during the chlorination process.</p>","PeriodicalId":148,"journal":{"name":"ChemPlusChem","volume":"90 10","pages":""},"PeriodicalIF":2.8000,"publicationDate":"2025-08-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Unveiling SalL Chlorinase Reaction Mechanism and Selectivity through Quantum Mechanical/Molecular Mechanics and Activation-Strain Model\",\"authors\":\"Néstor Gutiérrez-Sánchez, Fernando Mendizábal, Sebastián Miranda-Rojas\",\"doi\":\"10.1002/cplu.202500344\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Here, an exhaustive exploration of the reaction mechanism toward the chlorination process carried out by SalL, a chlorinase enzyme that catalyzes the conversion of SAM into 5′-chloro-5′-deoxyadenosine through an <i>S</i><sub><i>N</i></sub>2 reaction, is presented. To this end, molecular dynamics simulations and quantum mechanical/molecular mechanics calculations are performed, and 14 density functionals are benchmarked. Among the tested functionals, TPSSh(BJ) provides the closest energy barrier to experimental value. Three configurations of interaction between chloride and the halogen pocket are found, where the best model exhibits a barrier height of 20.1 kcal mol<sup>−1</sup>, close to the 19.9 kcal mol<sup>−1</sup> experimentally obtained. This model is characterized by the chloride interacting with the backbone-amide of Gly131 and Tyr130. The reaction pathway is calculated through the intrinsic reaction coordinate approach, and it is characterized using reaction force analysis and the activation-strain model with energy decomposition analysis to obtain chemical insights into the inner working of this enzyme. According to the main findings, the overstabilization of the halogen binding on the active site increases the barrier height, explaining the lack of activity against fluoride, while the interaction energy between nucleophile−electrophile is responsible of reducing the barrier height, with the orbital interaction energy as the main stabilizing factor during the chlorination process.</p>\",\"PeriodicalId\":148,\"journal\":{\"name\":\"ChemPlusChem\",\"volume\":\"90 10\",\"pages\":\"\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-08-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"ChemPlusChem\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cplu.202500344\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"ChemPlusChem","FirstCategoryId":"92","ListUrlMain":"https://chemistry-europe.onlinelibrary.wiley.com/doi/10.1002/cplu.202500344","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Unveiling SalL Chlorinase Reaction Mechanism and Selectivity through Quantum Mechanical/Molecular Mechanics and Activation-Strain Model
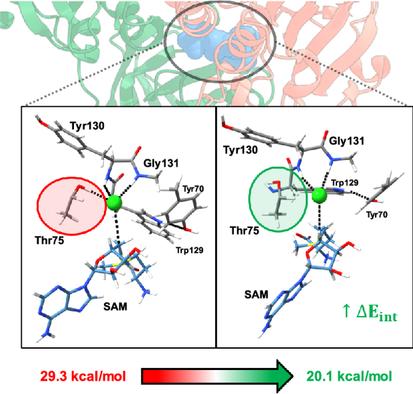
Here, an exhaustive exploration of the reaction mechanism toward the chlorination process carried out by SalL, a chlorinase enzyme that catalyzes the conversion of SAM into 5′-chloro-5′-deoxyadenosine through an SN2 reaction, is presented. To this end, molecular dynamics simulations and quantum mechanical/molecular mechanics calculations are performed, and 14 density functionals are benchmarked. Among the tested functionals, TPSSh(BJ) provides the closest energy barrier to experimental value. Three configurations of interaction between chloride and the halogen pocket are found, where the best model exhibits a barrier height of 20.1 kcal mol−1, close to the 19.9 kcal mol−1 experimentally obtained. This model is characterized by the chloride interacting with the backbone-amide of Gly131 and Tyr130. The reaction pathway is calculated through the intrinsic reaction coordinate approach, and it is characterized using reaction force analysis and the activation-strain model with energy decomposition analysis to obtain chemical insights into the inner working of this enzyme. According to the main findings, the overstabilization of the halogen binding on the active site increases the barrier height, explaining the lack of activity against fluoride, while the interaction energy between nucleophile−electrophile is responsible of reducing the barrier height, with the orbital interaction energy as the main stabilizing factor during the chlorination process.
期刊介绍:
ChemPlusChem is a peer-reviewed, general chemistry journal that brings readers the very best in multidisciplinary research centering on chemistry. It is published on behalf of Chemistry Europe, an association of 16 European chemical societies.
Fully comprehensive in its scope, ChemPlusChem publishes articles covering new results from at least two different aspects (subfields) of chemistry or one of chemistry and one of another scientific discipline (one chemistry topic plus another one, hence the title ChemPlusChem). All suitable submissions undergo balanced peer review by experts in the field to ensure the highest quality, originality, relevance, significance, and validity.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


