Qiaohong Wang, Ruhee D’Cunha, Abhishek Mitra, Stephen K. Gray*, Matthew Otten* and Laura Gagliardi*,
{"title":"局域活动空间法的非酉变分量子特征解及代价降低。","authors":"Qiaohong Wang, Ruhee D’Cunha, Abhishek Mitra, Stephen K. Gray*, Matthew Otten* and Laura Gagliardi*, ","doi":"10.1021/acs.jpca.5c03028","DOIUrl":null,"url":null,"abstract":"<p >Accurately describing strongly correlated systems with affordable quantum resources remains a central challenge for quantum chemistry applications on near and intermediate term quantum computers. The localized active space self-consistent field (LASSCF) approximates the complete active space self-consistent field (CASSCF) by generating active space-based wave functions within specific fragments while treating interfragment correlation with mean-field approach, hence is computationally less expensive. Hardware-efficient ansatzes (HEA) offer affordable and shallower circuits, yet they often fail to capture the necessary correlation. Previously, Jastrow-factor-inspired nonunitary qubit operators were proposed to use with HEA for variational quantum eigensolver (VQE) calculations (so-called nuVQE), as they do not increase circuit depths and recover correlation beyond the mean-field level for Hartree–Fock initial states. Here, we explore running nuVQE with LASSCF as the initial state. The method, named LAS-nuVQE, is shown to recover interfragment correlations, reach chemical accuracy with a small number of gates (<70) in both H<sub>4</sub> and square cyclobutadiene (C<sub>4</sub>H<sub>4</sub>), and produces more accurate energetics than its HEA counterparts at all circuit depths. To further address the inherent symmetry-breaking in HEA, we implemented spin-constrained LAS-nuVQE to extend the capabilities of HEA further and show spin-pure results for square cyclobutadiene. We also mitigate the increased measurement overhead of nuVQE via Pauli grouping and shot-frugal sampling, reducing measurement costs by up to 2 orders of magnitude compared to ungrouped operator, and show that one can achieve better accuracy with a small number of shots (10<sup>3–4</sup>) per one expectation value calculation compared to noiseless simulations with one or two orders of magnitude more shots. Finally, wall clock time estimates show that, with our measurement mitigation protocols, nuVQE becomes a cheaper and more accurate alternative than vanilla VQE with HEA. Taken together, these developments illustrate a practical pathway toward performing multireference chemical simulations with accuracy and affordable resources on today’s quantum hardware, achieving both accuracy and affordability in challenging correlated systems.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"129 34","pages":"7999–8012"},"PeriodicalIF":2.8000,"publicationDate":"2025-08-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Nonunitary Variational Quantum Eigensolver with the Localized Active Space Method and Cost Mitigation\",\"authors\":\"Qiaohong Wang, Ruhee D’Cunha, Abhishek Mitra, Stephen K. Gray*, Matthew Otten* and Laura Gagliardi*, \",\"doi\":\"10.1021/acs.jpca.5c03028\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Accurately describing strongly correlated systems with affordable quantum resources remains a central challenge for quantum chemistry applications on near and intermediate term quantum computers. The localized active space self-consistent field (LASSCF) approximates the complete active space self-consistent field (CASSCF) by generating active space-based wave functions within specific fragments while treating interfragment correlation with mean-field approach, hence is computationally less expensive. Hardware-efficient ansatzes (HEA) offer affordable and shallower circuits, yet they often fail to capture the necessary correlation. Previously, Jastrow-factor-inspired nonunitary qubit operators were proposed to use with HEA for variational quantum eigensolver (VQE) calculations (so-called nuVQE), as they do not increase circuit depths and recover correlation beyond the mean-field level for Hartree–Fock initial states. Here, we explore running nuVQE with LASSCF as the initial state. The method, named LAS-nuVQE, is shown to recover interfragment correlations, reach chemical accuracy with a small number of gates (<70) in both H<sub>4</sub> and square cyclobutadiene (C<sub>4</sub>H<sub>4</sub>), and produces more accurate energetics than its HEA counterparts at all circuit depths. To further address the inherent symmetry-breaking in HEA, we implemented spin-constrained LAS-nuVQE to extend the capabilities of HEA further and show spin-pure results for square cyclobutadiene. We also mitigate the increased measurement overhead of nuVQE via Pauli grouping and shot-frugal sampling, reducing measurement costs by up to 2 orders of magnitude compared to ungrouped operator, and show that one can achieve better accuracy with a small number of shots (10<sup>3–4</sup>) per one expectation value calculation compared to noiseless simulations with one or two orders of magnitude more shots. Finally, wall clock time estimates show that, with our measurement mitigation protocols, nuVQE becomes a cheaper and more accurate alternative than vanilla VQE with HEA. Taken together, these developments illustrate a practical pathway toward performing multireference chemical simulations with accuracy and affordable resources on today’s quantum hardware, achieving both accuracy and affordability in challenging correlated systems.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"129 34\",\"pages\":\"7999–8012\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-08-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.5c03028\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.5c03028","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Nonunitary Variational Quantum Eigensolver with the Localized Active Space Method and Cost Mitigation
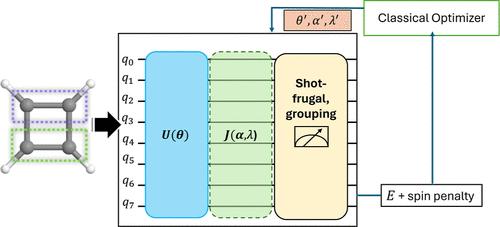
Accurately describing strongly correlated systems with affordable quantum resources remains a central challenge for quantum chemistry applications on near and intermediate term quantum computers. The localized active space self-consistent field (LASSCF) approximates the complete active space self-consistent field (CASSCF) by generating active space-based wave functions within specific fragments while treating interfragment correlation with mean-field approach, hence is computationally less expensive. Hardware-efficient ansatzes (HEA) offer affordable and shallower circuits, yet they often fail to capture the necessary correlation. Previously, Jastrow-factor-inspired nonunitary qubit operators were proposed to use with HEA for variational quantum eigensolver (VQE) calculations (so-called nuVQE), as they do not increase circuit depths and recover correlation beyond the mean-field level for Hartree–Fock initial states. Here, we explore running nuVQE with LASSCF as the initial state. The method, named LAS-nuVQE, is shown to recover interfragment correlations, reach chemical accuracy with a small number of gates (<70) in both H4 and square cyclobutadiene (C4H4), and produces more accurate energetics than its HEA counterparts at all circuit depths. To further address the inherent symmetry-breaking in HEA, we implemented spin-constrained LAS-nuVQE to extend the capabilities of HEA further and show spin-pure results for square cyclobutadiene. We also mitigate the increased measurement overhead of nuVQE via Pauli grouping and shot-frugal sampling, reducing measurement costs by up to 2 orders of magnitude compared to ungrouped operator, and show that one can achieve better accuracy with a small number of shots (103–4) per one expectation value calculation compared to noiseless simulations with one or two orders of magnitude more shots. Finally, wall clock time estimates show that, with our measurement mitigation protocols, nuVQE becomes a cheaper and more accurate alternative than vanilla VQE with HEA. Taken together, these developments illustrate a practical pathway toward performing multireference chemical simulations with accuracy and affordable resources on today’s quantum hardware, achieving both accuracy and affordability in challenging correlated systems.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


