Jiaxin Xie, Mengmeng Jia, Xuan Ren, Siyu Cheng, Shuyuan Liu, Yiwen Hu, Song Cheng, Yun Hin Taufiq-Yap and Yang Li*,
{"title":"硝酸2-乙基己基的热化学和速率动力学研究:H原子抽离反应方法。","authors":"Jiaxin Xie, Mengmeng Jia, Xuan Ren, Siyu Cheng, Shuyuan Liu, Yiwen Hu, Song Cheng, Yun Hin Taufiq-Yap and Yang Li*, ","doi":"10.1021/acs.jpca.5c03710","DOIUrl":null,"url":null,"abstract":"<p >2-Ethylhexyl nitrate (EHN) is a promising high energy liquid fuel candidate due to its high reactivity and rapid energy release. Understanding the multichannel H atom abstraction mechanisms in EHN combustion is essential for improving combustion modeling accuracy. This study employs <i>ab initio</i> methods and transition state theory (TST) to systematically investigate H-abstraction reactions at four specific sites in EHN, initiated by six abstractors: Ḣ, ȮH, HȮ<sub>2</sub>, ṄO<sub>2</sub>, O<sub>2</sub>, and ĊN. Geometry optimizations, frequency analyses, and dihedral scans were performed using the M06-2X/6-311++G (d, p) method. Torsional modes were treated by using a one-dimensional hindered rotor approximation. Single-point energies (SPEs) were calculated via QCISD/cc-pVXZ (X = T, Q) and MP2/cc-pVYZ (Y = D, T, and Q) with complete basis set (CBS) extrapolation. High-pressure limit rate constants of all of the reaction channels were calculated at a wide temperature range from 298.15 to 2000 K by taking asymmetric Eckart tunneling corrections into account. In addition, the bond dissociation energies (BDEs) of seven bonds in EHN were determined, and thermochemical properties of relevant species were calculated via the atomization method. Finally, the effect of different functional groups on the reactivity of H abstraction was explored. Key findings include: (1) an O–N bond, with the lowest BDE at 42.83 kcal mol<sup>–1</sup>, demonstrating that it is the most susceptible to bond fission; (2) C<sub>2</sub> is the most reactive site for H-abstraction; (3) ĊN and ȮH are particularly competitive abstractors; and (4) nitrogen-containing functional groups have a very slight effect on the H abstraction reaction as compared to non-nitrogen-containing functional groups.</p>","PeriodicalId":59,"journal":{"name":"The Journal of Physical Chemistry A","volume":"129 34","pages":"7927–7938"},"PeriodicalIF":2.8000,"publicationDate":"2025-08-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Elucidating the Thermochemistry and Rate Kinetics of 2-Ethylhexyl Nitrate: An H Atom Abstraction Reactions Approach\",\"authors\":\"Jiaxin Xie, Mengmeng Jia, Xuan Ren, Siyu Cheng, Shuyuan Liu, Yiwen Hu, Song Cheng, Yun Hin Taufiq-Yap and Yang Li*, \",\"doi\":\"10.1021/acs.jpca.5c03710\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >2-Ethylhexyl nitrate (EHN) is a promising high energy liquid fuel candidate due to its high reactivity and rapid energy release. Understanding the multichannel H atom abstraction mechanisms in EHN combustion is essential for improving combustion modeling accuracy. This study employs <i>ab initio</i> methods and transition state theory (TST) to systematically investigate H-abstraction reactions at four specific sites in EHN, initiated by six abstractors: Ḣ, ȮH, HȮ<sub>2</sub>, ṄO<sub>2</sub>, O<sub>2</sub>, and ĊN. Geometry optimizations, frequency analyses, and dihedral scans were performed using the M06-2X/6-311++G (d, p) method. Torsional modes were treated by using a one-dimensional hindered rotor approximation. Single-point energies (SPEs) were calculated via QCISD/cc-pVXZ (X = T, Q) and MP2/cc-pVYZ (Y = D, T, and Q) with complete basis set (CBS) extrapolation. High-pressure limit rate constants of all of the reaction channels were calculated at a wide temperature range from 298.15 to 2000 K by taking asymmetric Eckart tunneling corrections into account. In addition, the bond dissociation energies (BDEs) of seven bonds in EHN were determined, and thermochemical properties of relevant species were calculated via the atomization method. Finally, the effect of different functional groups on the reactivity of H abstraction was explored. Key findings include: (1) an O–N bond, with the lowest BDE at 42.83 kcal mol<sup>–1</sup>, demonstrating that it is the most susceptible to bond fission; (2) C<sub>2</sub> is the most reactive site for H-abstraction; (3) ĊN and ȮH are particularly competitive abstractors; and (4) nitrogen-containing functional groups have a very slight effect on the H abstraction reaction as compared to non-nitrogen-containing functional groups.</p>\",\"PeriodicalId\":59,\"journal\":{\"name\":\"The Journal of Physical Chemistry A\",\"volume\":\"129 34\",\"pages\":\"7927–7938\"},\"PeriodicalIF\":2.8000,\"publicationDate\":\"2025-08-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry A\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.jpca.5c03710\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry A","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpca.5c03710","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
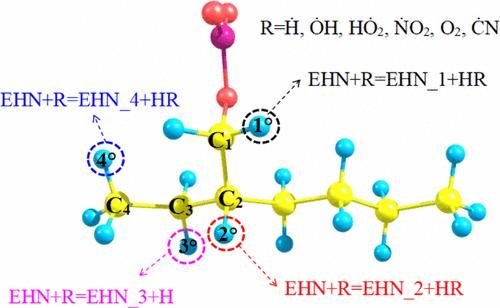
Elucidating the Thermochemistry and Rate Kinetics of 2-Ethylhexyl Nitrate: An H Atom Abstraction Reactions Approach
2-Ethylhexyl nitrate (EHN) is a promising high energy liquid fuel candidate due to its high reactivity and rapid energy release. Understanding the multichannel H atom abstraction mechanisms in EHN combustion is essential for improving combustion modeling accuracy. This study employs ab initio methods and transition state theory (TST) to systematically investigate H-abstraction reactions at four specific sites in EHN, initiated by six abstractors: Ḣ, ȮH, HȮ2, ṄO2, O2, and ĊN. Geometry optimizations, frequency analyses, and dihedral scans were performed using the M06-2X/6-311++G (d, p) method. Torsional modes were treated by using a one-dimensional hindered rotor approximation. Single-point energies (SPEs) were calculated via QCISD/cc-pVXZ (X = T, Q) and MP2/cc-pVYZ (Y = D, T, and Q) with complete basis set (CBS) extrapolation. High-pressure limit rate constants of all of the reaction channels were calculated at a wide temperature range from 298.15 to 2000 K by taking asymmetric Eckart tunneling corrections into account. In addition, the bond dissociation energies (BDEs) of seven bonds in EHN were determined, and thermochemical properties of relevant species were calculated via the atomization method. Finally, the effect of different functional groups on the reactivity of H abstraction was explored. Key findings include: (1) an O–N bond, with the lowest BDE at 42.83 kcal mol–1, demonstrating that it is the most susceptible to bond fission; (2) C2 is the most reactive site for H-abstraction; (3) ĊN and ȮH are particularly competitive abstractors; and (4) nitrogen-containing functional groups have a very slight effect on the H abstraction reaction as compared to non-nitrogen-containing functional groups.
期刊介绍:
The Journal of Physical Chemistry A is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


