D. V. Krivorotov, D. A. Belinskaia, A. S. Smirnov, V. V. Suslonov, N. V. Goncharov, V. A. Kuznetsov
{"title":"阿片拮抗剂的几何构型和电荷对其与受体结合的影响","authors":"D. V. Krivorotov, D. A. Belinskaia, A. S. Smirnov, V. V. Suslonov, N. V. Goncharov, V. A. Kuznetsov","doi":"10.1134/S1990747825700254","DOIUrl":null,"url":null,"abstract":"<p>The effect of the geometric configuration and charge of membrane opioid receptor (OR) agonists and antagonists on binding to μ, δ, and κ opioid receptors has been studied by molecular docking. For the docking procedure, three-dimensional structures of pharmaceutical preparations obtained by X-ray diffraction analysis (XRD) and available in the Cambridge Crystal Structures Database (CCDC), as well as their three-dimensional models constructed in a molecular editor, were used. The three-dimensional crystal structure of nalmefene, which is absent in the CCDC database, was first obtained in the presented study by XRD. Protonated and deprotonated forms of ligands were considered. The results of the study using morphine, codeine, naloxone, naltrexone, and nalmefene as examples showed that the method of obtaining three-dimensional geometric structures of opioid receptor ligands practically did not affect the calculated free energy of binding ΔG, which indicates the possibility of using ligand models constructed <i>in silico</i> in computational experiments. The protonation state of the ligand molecule, on the contrary, had a significant effect on free energy of binding to OR, which might affect the properties of this group of drugs with pH changes in the body. When considering the binding features of opioid enantiomers to the ligand-binding site of μ-opioid receptors, using morphine as an example, it was shown that (–)-morphine and (+)-morphine share a common site for the cationic group, and not for phenolic hydroxyl, as previously assumed. At the same time, studies have shown that molecular docking only partially made it possible to describe the pharmacological effect of analgesics and their antagonists. For some substances, such as codeine and synthetic (+)-morphine, the experiment <i>in silico</i> overestimated the effectiveness of the drug’s interaction with OR, and this requires continued improvement of the corresponding calculation methods and models.</p>","PeriodicalId":484,"journal":{"name":"Biochemistry (Moscow), Supplement Series A: Membrane and Cell Biology","volume":"19 3","pages":"303 - 317"},"PeriodicalIF":1.4000,"publicationDate":"2025-08-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"In Silico Evaluation of the Effect of Geometrical Configuration and Charge of Opioid Antagonists on Their Binding to Receptors\",\"authors\":\"D. V. Krivorotov, D. A. Belinskaia, A. S. Smirnov, V. V. Suslonov, N. V. Goncharov, V. A. Kuznetsov\",\"doi\":\"10.1134/S1990747825700254\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The effect of the geometric configuration and charge of membrane opioid receptor (OR) agonists and antagonists on binding to μ, δ, and κ opioid receptors has been studied by molecular docking. For the docking procedure, three-dimensional structures of pharmaceutical preparations obtained by X-ray diffraction analysis (XRD) and available in the Cambridge Crystal Structures Database (CCDC), as well as their three-dimensional models constructed in a molecular editor, were used. The three-dimensional crystal structure of nalmefene, which is absent in the CCDC database, was first obtained in the presented study by XRD. Protonated and deprotonated forms of ligands were considered. The results of the study using morphine, codeine, naloxone, naltrexone, and nalmefene as examples showed that the method of obtaining three-dimensional geometric structures of opioid receptor ligands practically did not affect the calculated free energy of binding ΔG, which indicates the possibility of using ligand models constructed <i>in silico</i> in computational experiments. The protonation state of the ligand molecule, on the contrary, had a significant effect on free energy of binding to OR, which might affect the properties of this group of drugs with pH changes in the body. When considering the binding features of opioid enantiomers to the ligand-binding site of μ-opioid receptors, using morphine as an example, it was shown that (–)-morphine and (+)-morphine share a common site for the cationic group, and not for phenolic hydroxyl, as previously assumed. At the same time, studies have shown that molecular docking only partially made it possible to describe the pharmacological effect of analgesics and their antagonists. For some substances, such as codeine and synthetic (+)-morphine, the experiment <i>in silico</i> overestimated the effectiveness of the drug’s interaction with OR, and this requires continued improvement of the corresponding calculation methods and models.</p>\",\"PeriodicalId\":484,\"journal\":{\"name\":\"Biochemistry (Moscow), Supplement Series A: Membrane and Cell Biology\",\"volume\":\"19 3\",\"pages\":\"303 - 317\"},\"PeriodicalIF\":1.4000,\"publicationDate\":\"2025-08-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biochemistry (Moscow), Supplement Series A: Membrane and Cell Biology\",\"FirstCategoryId\":\"2\",\"ListUrlMain\":\"https://link.springer.com/article/10.1134/S1990747825700254\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biochemistry (Moscow), Supplement Series A: Membrane and Cell Biology","FirstCategoryId":"2","ListUrlMain":"https://link.springer.com/article/10.1134/S1990747825700254","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
In Silico Evaluation of the Effect of Geometrical Configuration and Charge of Opioid Antagonists on Their Binding to Receptors
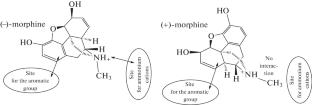
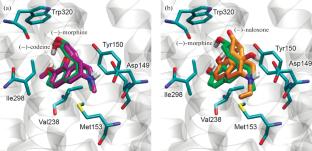
The effect of the geometric configuration and charge of membrane opioid receptor (OR) agonists and antagonists on binding to μ, δ, and κ opioid receptors has been studied by molecular docking. For the docking procedure, three-dimensional structures of pharmaceutical preparations obtained by X-ray diffraction analysis (XRD) and available in the Cambridge Crystal Structures Database (CCDC), as well as their three-dimensional models constructed in a molecular editor, were used. The three-dimensional crystal structure of nalmefene, which is absent in the CCDC database, was first obtained in the presented study by XRD. Protonated and deprotonated forms of ligands were considered. The results of the study using morphine, codeine, naloxone, naltrexone, and nalmefene as examples showed that the method of obtaining three-dimensional geometric structures of opioid receptor ligands practically did not affect the calculated free energy of binding ΔG, which indicates the possibility of using ligand models constructed in silico in computational experiments. The protonation state of the ligand molecule, on the contrary, had a significant effect on free energy of binding to OR, which might affect the properties of this group of drugs with pH changes in the body. When considering the binding features of opioid enantiomers to the ligand-binding site of μ-opioid receptors, using morphine as an example, it was shown that (–)-morphine and (+)-morphine share a common site for the cationic group, and not for phenolic hydroxyl, as previously assumed. At the same time, studies have shown that molecular docking only partially made it possible to describe the pharmacological effect of analgesics and their antagonists. For some substances, such as codeine and synthetic (+)-morphine, the experiment in silico overestimated the effectiveness of the drug’s interaction with OR, and this requires continued improvement of the corresponding calculation methods and models.
期刊介绍:
Biochemistry (Moscow), Supplement Series A: Membrane and Cell Biology is an international peer reviewed journal that publishes original articles on physical, chemical, and molecular mechanisms that underlie basic properties of biological membranes and mediate membrane-related cellular functions. The primary topics of the journal are membrane structure, mechanisms of membrane transport, bioenergetics and photobiology, intracellular signaling as well as membrane aspects of cell biology, immunology, and medicine. The journal is multidisciplinary and gives preference to those articles that employ a variety of experimental approaches, basically in biophysics but also in biochemistry, cytology, and molecular biology. The journal publishes articles that strive for unveiling membrane and cellular functions through innovative theoretical models and computer simulations.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


