Tamiris Nogueira Bezerra Bueno, Társis Paiva Vieira, Vera Lúcia Gil-da-Silva-Lopes
{"title":"唇腭裂的基因检测:基于单一巴西公共遗传学服务的反思。","authors":"Tamiris Nogueira Bezerra Bueno, Társis Paiva Vieira, Vera Lúcia Gil-da-Silva-Lopes","doi":"10.1186/s13023-025-03967-y","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Genomic medicine has allowed for an improvement in the diagnosis and molecular understanding of congenital defects. However, its implementation into routine clinical practice demands enormous challenges worldwide. This study describes the etiological diversity and access to genetic diagnosis of individuals with oral clefts (OC) at a single genetics service.</p><p><strong>Results: </strong>This cross-sectional and descriptive study analyzed primary records of the Brazilian Database on Craniofacial Anomalies from 2006 to 2019, before the National Policy of Comprehensive Care for People with Rare Diseases (NPCCPRD) implementation in this service. Among 103 individuals (51 Female and 52 Male), the proportion of syndromic OC (SOC) and non-syndromic OC (NSOC) was 73.8% and 26.2%, respectively, showing that NSOC seems not to be referred for genetic evaluation. Diagnosis occurred in 64/103 (62.13%) cases; 36/64 (56,25%) had clinical diagnoses, of which 27/36 were NSOC. The tests allowing a conclusive diagnosis were whole exome sequencing (WES) (11/20-55.00%), followed by chromosomal microarray analysis (CMA) (4/52-7.69%), Fluorescent in situ hybridization (FISH) (6/21-28.57%), multiplex ligation-dependent probe amplification (MLPA) (2/32-6.25%), and G-banding karyotype (6/72-8.33%). Age at diagnosis ranged from 0 to 46 years (mean = 9.56 /median = 7). Diagnostic investigations of 39/76 SOC cases are still ongoing, relying on clinical follow-up and genetic tests.</p><p><strong>Conclusions: </strong>Etiological diversity reinforces the need for different laboratory resources and clinical follow-up. These results and reflections about the need to implement Genomic Medicine are of universal interest. They also show the need to improve public health policies for genetic evaluation, diagnostic tests, and genetic counseling for an effective NPCCPRD.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"435"},"PeriodicalIF":3.5000,"publicationDate":"2025-08-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12358060/pdf/","citationCount":"0","resultStr":"{\"title\":\"Genetic testing for oral clefts: reflections based on a single Brazilian public genetics service.\",\"authors\":\"Tamiris Nogueira Bezerra Bueno, Társis Paiva Vieira, Vera Lúcia Gil-da-Silva-Lopes\",\"doi\":\"10.1186/s13023-025-03967-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Genomic medicine has allowed for an improvement in the diagnosis and molecular understanding of congenital defects. However, its implementation into routine clinical practice demands enormous challenges worldwide. This study describes the etiological diversity and access to genetic diagnosis of individuals with oral clefts (OC) at a single genetics service.</p><p><strong>Results: </strong>This cross-sectional and descriptive study analyzed primary records of the Brazilian Database on Craniofacial Anomalies from 2006 to 2019, before the National Policy of Comprehensive Care for People with Rare Diseases (NPCCPRD) implementation in this service. Among 103 individuals (51 Female and 52 Male), the proportion of syndromic OC (SOC) and non-syndromic OC (NSOC) was 73.8% and 26.2%, respectively, showing that NSOC seems not to be referred for genetic evaluation. Diagnosis occurred in 64/103 (62.13%) cases; 36/64 (56,25%) had clinical diagnoses, of which 27/36 were NSOC. The tests allowing a conclusive diagnosis were whole exome sequencing (WES) (11/20-55.00%), followed by chromosomal microarray analysis (CMA) (4/52-7.69%), Fluorescent in situ hybridization (FISH) (6/21-28.57%), multiplex ligation-dependent probe amplification (MLPA) (2/32-6.25%), and G-banding karyotype (6/72-8.33%). Age at diagnosis ranged from 0 to 46 years (mean = 9.56 /median = 7). Diagnostic investigations of 39/76 SOC cases are still ongoing, relying on clinical follow-up and genetic tests.</p><p><strong>Conclusions: </strong>Etiological diversity reinforces the need for different laboratory resources and clinical follow-up. These results and reflections about the need to implement Genomic Medicine are of universal interest. They also show the need to improve public health policies for genetic evaluation, diagnostic tests, and genetic counseling for an effective NPCCPRD.</p>\",\"PeriodicalId\":19651,\"journal\":{\"name\":\"Orphanet Journal of Rare Diseases\",\"volume\":\"20 1\",\"pages\":\"435\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2025-08-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12358060/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Orphanet Journal of Rare Diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13023-025-03967-y\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-03967-y","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Genetic testing for oral clefts: reflections based on a single Brazilian public genetics service.
Background: Genomic medicine has allowed for an improvement in the diagnosis and molecular understanding of congenital defects. However, its implementation into routine clinical practice demands enormous challenges worldwide. This study describes the etiological diversity and access to genetic diagnosis of individuals with oral clefts (OC) at a single genetics service.
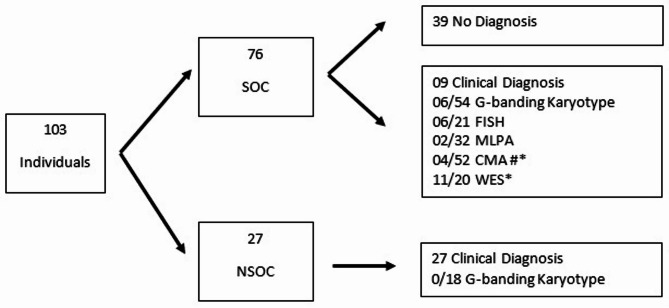
Results: This cross-sectional and descriptive study analyzed primary records of the Brazilian Database on Craniofacial Anomalies from 2006 to 2019, before the National Policy of Comprehensive Care for People with Rare Diseases (NPCCPRD) implementation in this service. Among 103 individuals (51 Female and 52 Male), the proportion of syndromic OC (SOC) and non-syndromic OC (NSOC) was 73.8% and 26.2%, respectively, showing that NSOC seems not to be referred for genetic evaluation. Diagnosis occurred in 64/103 (62.13%) cases; 36/64 (56,25%) had clinical diagnoses, of which 27/36 were NSOC. The tests allowing a conclusive diagnosis were whole exome sequencing (WES) (11/20-55.00%), followed by chromosomal microarray analysis (CMA) (4/52-7.69%), Fluorescent in situ hybridization (FISH) (6/21-28.57%), multiplex ligation-dependent probe amplification (MLPA) (2/32-6.25%), and G-banding karyotype (6/72-8.33%). Age at diagnosis ranged from 0 to 46 years (mean = 9.56 /median = 7). Diagnostic investigations of 39/76 SOC cases are still ongoing, relying on clinical follow-up and genetic tests.
Conclusions: Etiological diversity reinforces the need for different laboratory resources and clinical follow-up. These results and reflections about the need to implement Genomic Medicine are of universal interest. They also show the need to improve public health policies for genetic evaluation, diagnostic tests, and genetic counseling for an effective NPCCPRD.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


