Xinyu Liu, Zhen Zhang, Chao Tan, Yinquan Ai, Hao Liu, Yuan Li, Jin Yang, Yongyan Song
{"title":"单细胞转录组学研究中机器学习应用的全球趋势。","authors":"Xinyu Liu, Zhen Zhang, Chao Tan, Yinquan Ai, Hao Liu, Yuan Li, Jin Yang, Yongyan Song","doi":"10.1186/s41065-025-00528-y","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Single-cell RNA sequencing (scRNA-seq) has revolutionized cellular heterogeneity analysis by decoding gene expression profiles at individual cell level, while machine learning (ML) has emerged as core computational tool for clustering analysis, dimensionality reduction modeling and developmental trajectory inference in single-cell transcriptomics(SCT). Although 3,307 papers have been published in past two decades, there remains lack of bibliometric review comprehensively addressing methodological evolution, technical challenges and clinical translation pathways. This study aims to fill research gap through bibliometric and visual analysis, revealing technological evolution trends and future development directions.</p><p><strong>Methods: </strong>Using 3,307 publications from Web of Science Core Collection(WOSCC), we conducted bibliometric and visualization analysis through CiteSpace and VOSviewer to systematically review research trends, national/institutional contributions, keyword co-occurrence networks and co-citation relationships. Data screening strictly limited to English articles and reviews, excluding irrelevant document types, focusing on core application scenarios of ML in SCT.</p><p><strong>Results: </strong>China and United States dominated research output (combined 65%), with China leading in publication volume (54.8%) while US demonstrating academic influence through H-index 84 and 37,135 total citations. Research hotspots concentrated on random forest (RF) and deep learning models, showing transition from algorithm development to clinical applications (e.g., tumor immune microenvironment analysis). Chinese Academy of Sciences and Harvard University emerged as core collaboration hubs, with international cooperation network primarily featuring US-China collaboration. Keyword clustering revealed four themes: gene expression, immunotherapy, bioinformatics, and inflammation-related research. Technical bottlenecks included data heterogeneity, insufficient model interpretability and weak cross-dataset generalization capability.</p><p><strong>Conclusion: </strong>ML-scRNA-seq integration has advanced cellular heterogeneity analysis and precision medicine development. Future directions should optimize deep learning architectures, enhance model generalization capabilities, and promote technical translation through multi-omics and clinical data integration. Interdisciplinary collaboration represents key to overcoming current limitations (e.g., data standardization, algorithm interpretability), ultimately realizing deep integration between single-cell technologies and precision medicine.</p>","PeriodicalId":12862,"journal":{"name":"Hereditas","volume":"162 1","pages":"164"},"PeriodicalIF":2.5000,"publicationDate":"2025-08-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12357469/pdf/","citationCount":"0","resultStr":"{\"title\":\"Global trends in machine learning applications for single-cell transcriptomics research.\",\"authors\":\"Xinyu Liu, Zhen Zhang, Chao Tan, Yinquan Ai, Hao Liu, Yuan Li, Jin Yang, Yongyan Song\",\"doi\":\"10.1186/s41065-025-00528-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Single-cell RNA sequencing (scRNA-seq) has revolutionized cellular heterogeneity analysis by decoding gene expression profiles at individual cell level, while machine learning (ML) has emerged as core computational tool for clustering analysis, dimensionality reduction modeling and developmental trajectory inference in single-cell transcriptomics(SCT). Although 3,307 papers have been published in past two decades, there remains lack of bibliometric review comprehensively addressing methodological evolution, technical challenges and clinical translation pathways. This study aims to fill research gap through bibliometric and visual analysis, revealing technological evolution trends and future development directions.</p><p><strong>Methods: </strong>Using 3,307 publications from Web of Science Core Collection(WOSCC), we conducted bibliometric and visualization analysis through CiteSpace and VOSviewer to systematically review research trends, national/institutional contributions, keyword co-occurrence networks and co-citation relationships. Data screening strictly limited to English articles and reviews, excluding irrelevant document types, focusing on core application scenarios of ML in SCT.</p><p><strong>Results: </strong>China and United States dominated research output (combined 65%), with China leading in publication volume (54.8%) while US demonstrating academic influence through H-index 84 and 37,135 total citations. Research hotspots concentrated on random forest (RF) and deep learning models, showing transition from algorithm development to clinical applications (e.g., tumor immune microenvironment analysis). Chinese Academy of Sciences and Harvard University emerged as core collaboration hubs, with international cooperation network primarily featuring US-China collaboration. Keyword clustering revealed four themes: gene expression, immunotherapy, bioinformatics, and inflammation-related research. Technical bottlenecks included data heterogeneity, insufficient model interpretability and weak cross-dataset generalization capability.</p><p><strong>Conclusion: </strong>ML-scRNA-seq integration has advanced cellular heterogeneity analysis and precision medicine development. Future directions should optimize deep learning architectures, enhance model generalization capabilities, and promote technical translation through multi-omics and clinical data integration. Interdisciplinary collaboration represents key to overcoming current limitations (e.g., data standardization, algorithm interpretability), ultimately realizing deep integration between single-cell technologies and precision medicine.</p>\",\"PeriodicalId\":12862,\"journal\":{\"name\":\"Hereditas\",\"volume\":\"162 1\",\"pages\":\"164\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2025-08-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12357469/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Hereditas\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1186/s41065-025-00528-y\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Hereditas","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s41065-025-00528-y","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
摘要
背景:单细胞RNA测序(scRNA-seq)通过在单个细胞水平上解码基因表达谱,彻底改变了细胞异质性分析,而机器学习(ML)已成为单细胞转录组学(SCT)中聚类分析、降维建模和发育轨迹推断的核心计算工具。尽管在过去的二十年中已经发表了3307篇论文,但仍然缺乏全面解决方法演变,技术挑战和临床翻译途径的文献计量学综述。本研究旨在通过文献计量和可视化分析来填补研究空白,揭示技术演进趋势和未来发展方向。方法:利用Web of Science Core Collection(WOSCC)的3307篇论文,通过CiteSpace和VOSviewer进行文献计量和可视化分析,系统回顾研究趋势、国家/机构贡献、关键词共现网络和共被引关系。数据筛选严格限于英文文章和综述,排除不相关的文档类型,重点关注ML在SCT中的核心应用场景。结果:中美两国的研究产出占主导地位(合计65%),其中中国的发表量领先(54.8%),美国的h指数为84,总引用数为37135。研究热点集中在随机森林(random forest, RF)和深度学习模型,呈现出从算法开发到临床应用(如肿瘤免疫微环境分析)的过渡。中国科学院和哈佛大学成为核心合作中心,形成以中美合作为主的国际合作网络。关键词聚类揭示了四个主题:基因表达、免疫治疗、生物信息学和炎症相关研究。技术瓶颈包括数据异构性、模型可解释性不足和跨数据集泛化能力弱。结论:ML-scRNA-seq整合具有促进细胞异质性分析和精准医学发展的作用。未来的发展方向应该是优化深度学习架构,增强模型泛化能力,并通过多组学和临床数据整合促进技术转化。跨学科合作是克服当前限制(例如,数据标准化,算法可解释性)的关键,最终实现单细胞技术与精准医疗之间的深度集成。
Global trends in machine learning applications for single-cell transcriptomics research.
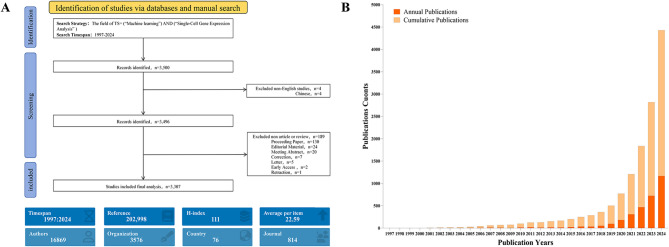
Background: Single-cell RNA sequencing (scRNA-seq) has revolutionized cellular heterogeneity analysis by decoding gene expression profiles at individual cell level, while machine learning (ML) has emerged as core computational tool for clustering analysis, dimensionality reduction modeling and developmental trajectory inference in single-cell transcriptomics(SCT). Although 3,307 papers have been published in past two decades, there remains lack of bibliometric review comprehensively addressing methodological evolution, technical challenges and clinical translation pathways. This study aims to fill research gap through bibliometric and visual analysis, revealing technological evolution trends and future development directions.
Methods: Using 3,307 publications from Web of Science Core Collection(WOSCC), we conducted bibliometric and visualization analysis through CiteSpace and VOSviewer to systematically review research trends, national/institutional contributions, keyword co-occurrence networks and co-citation relationships. Data screening strictly limited to English articles and reviews, excluding irrelevant document types, focusing on core application scenarios of ML in SCT.
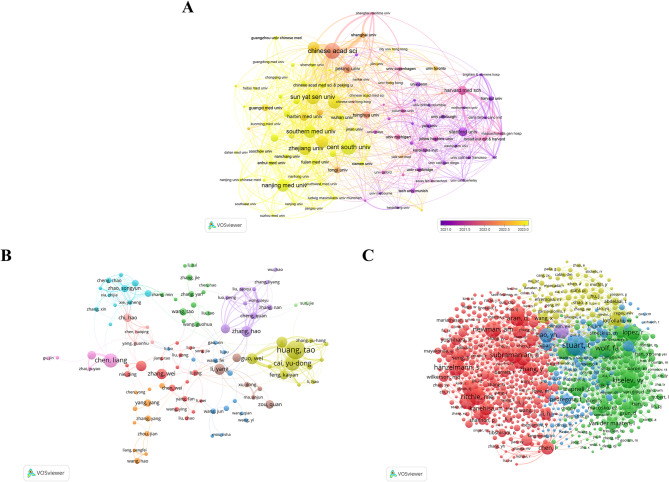
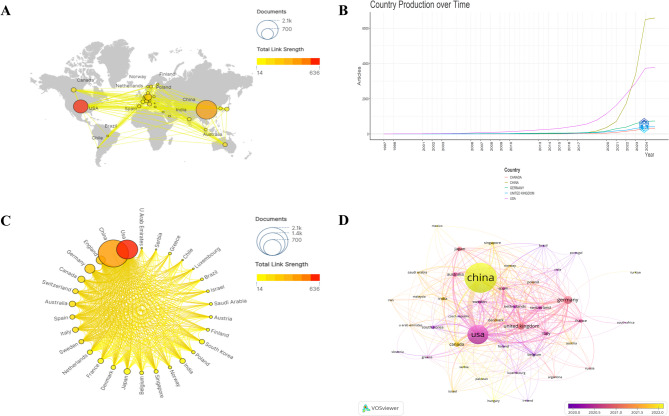
Results: China and United States dominated research output (combined 65%), with China leading in publication volume (54.8%) while US demonstrating academic influence through H-index 84 and 37,135 total citations. Research hotspots concentrated on random forest (RF) and deep learning models, showing transition from algorithm development to clinical applications (e.g., tumor immune microenvironment analysis). Chinese Academy of Sciences and Harvard University emerged as core collaboration hubs, with international cooperation network primarily featuring US-China collaboration. Keyword clustering revealed four themes: gene expression, immunotherapy, bioinformatics, and inflammation-related research. Technical bottlenecks included data heterogeneity, insufficient model interpretability and weak cross-dataset generalization capability.
Conclusion: ML-scRNA-seq integration has advanced cellular heterogeneity analysis and precision medicine development. Future directions should optimize deep learning architectures, enhance model generalization capabilities, and promote technical translation through multi-omics and clinical data integration. Interdisciplinary collaboration represents key to overcoming current limitations (e.g., data standardization, algorithm interpretability), ultimately realizing deep integration between single-cell technologies and precision medicine.
HereditasBiochemistry, Genetics and Molecular Biology-Genetics
CiteScore
3.80
自引率
3.70%
发文量
0
期刊介绍:
For almost a century, Hereditas has published original cutting-edge research and reviews. As the Official journal of the Mendelian Society of Lund, the journal welcomes research from across all areas of genetics and genomics. Topics of interest include human and medical genetics, animal and plant genetics, microbial genetics, agriculture and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


