Cristian Camilo Gaviria-Sabogal, Ingrid Tatyana Bernal, Yasmín Sánchez-Gómez, William Usaquén, Andrea Casas-Vargas, Nora Contreras Bravo, Adrien Morel, Dora J Fonseca-Mendoza, Carlos M Restrepo, Rodrigo Cabrera
{"title":"通过从全外显子组测序数据中推断IBD和亲属关系,在孤立的安第斯社区中改进罕见疾病变异发现。","authors":"Cristian Camilo Gaviria-Sabogal, Ingrid Tatyana Bernal, Yasmín Sánchez-Gómez, William Usaquén, Andrea Casas-Vargas, Nora Contreras Bravo, Adrien Morel, Dora J Fonseca-Mendoza, Carlos M Restrepo, Rodrigo Cabrera","doi":"10.1093/hmg/ddaf132","DOIUrl":null,"url":null,"abstract":"<p><p>Rare genetic diseases pose significant diagnostic challenges, especially in geographically isolated populations where consanguinity, founder effects, and novel variants often influence disease patterns. Whole-exome sequencing (WES) is standard practice for rare disease diagnostics, but its limited coverage of noncoding regions limits inheritance-by-descent (IBD) and Runs of Homozygosity (RoH) inference. In this study, we tested an imputation-enhanced IBD and RoH detection method using WES data of 84 individuals from 51 families in Boyacá, Colombia-an Andean region with complex admixed American ancestry. By leveraging large, multi-ancestry reference panels to impute genotypes and increase variant distribution, we achieved improved detection of IBD and RoH regions, with KING showing the best results among the different tools that were tested. Imputation with the 1000 Genome reference panel underperformed compared to raw WES data, whereas large reference panels with diverse ancestry showed the best performance. By integrating these refined IBD results with pedigree information, we identified cryptic family relationships, clarified the role of consanguinity, and improved the prioritization of candidate variants. Our findings show that imputation-enhanced IBD analyses can bolster the utility of WES for rare disease studies, contributing to more accurate and timely genetic diagnoses.</p>","PeriodicalId":13070,"journal":{"name":"Human molecular genetics","volume":" ","pages":"1733-1743"},"PeriodicalIF":3.2000,"publicationDate":"2025-10-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12498279/pdf/","citationCount":"0","resultStr":"{\"title\":\"Refining rare disease variant discovery in an isolated Andean community through imputation-enhanced IBD and kinship inference from whole exome sequencing data.\",\"authors\":\"Cristian Camilo Gaviria-Sabogal, Ingrid Tatyana Bernal, Yasmín Sánchez-Gómez, William Usaquén, Andrea Casas-Vargas, Nora Contreras Bravo, Adrien Morel, Dora J Fonseca-Mendoza, Carlos M Restrepo, Rodrigo Cabrera\",\"doi\":\"10.1093/hmg/ddaf132\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Rare genetic diseases pose significant diagnostic challenges, especially in geographically isolated populations where consanguinity, founder effects, and novel variants often influence disease patterns. Whole-exome sequencing (WES) is standard practice for rare disease diagnostics, but its limited coverage of noncoding regions limits inheritance-by-descent (IBD) and Runs of Homozygosity (RoH) inference. In this study, we tested an imputation-enhanced IBD and RoH detection method using WES data of 84 individuals from 51 families in Boyacá, Colombia-an Andean region with complex admixed American ancestry. By leveraging large, multi-ancestry reference panels to impute genotypes and increase variant distribution, we achieved improved detection of IBD and RoH regions, with KING showing the best results among the different tools that were tested. Imputation with the 1000 Genome reference panel underperformed compared to raw WES data, whereas large reference panels with diverse ancestry showed the best performance. By integrating these refined IBD results with pedigree information, we identified cryptic family relationships, clarified the role of consanguinity, and improved the prioritization of candidate variants. Our findings show that imputation-enhanced IBD analyses can bolster the utility of WES for rare disease studies, contributing to more accurate and timely genetic diagnoses.</p>\",\"PeriodicalId\":13070,\"journal\":{\"name\":\"Human molecular genetics\",\"volume\":\" \",\"pages\":\"1733-1743\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2025-10-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12498279/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Human molecular genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/hmg/ddaf132\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human molecular genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/hmg/ddaf132","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Refining rare disease variant discovery in an isolated Andean community through imputation-enhanced IBD and kinship inference from whole exome sequencing data.
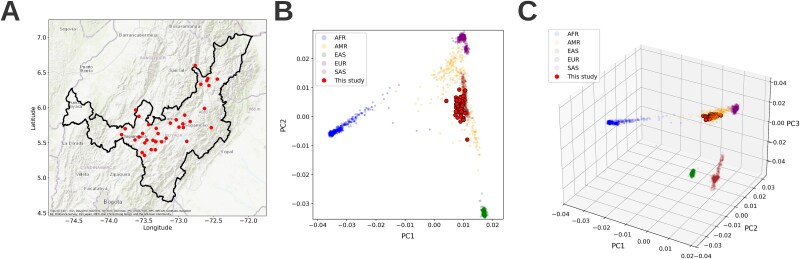
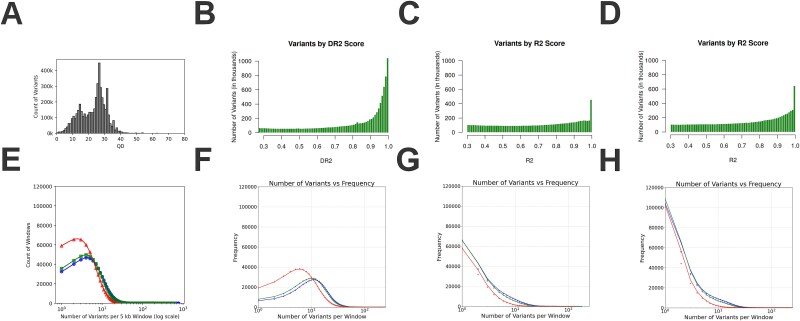
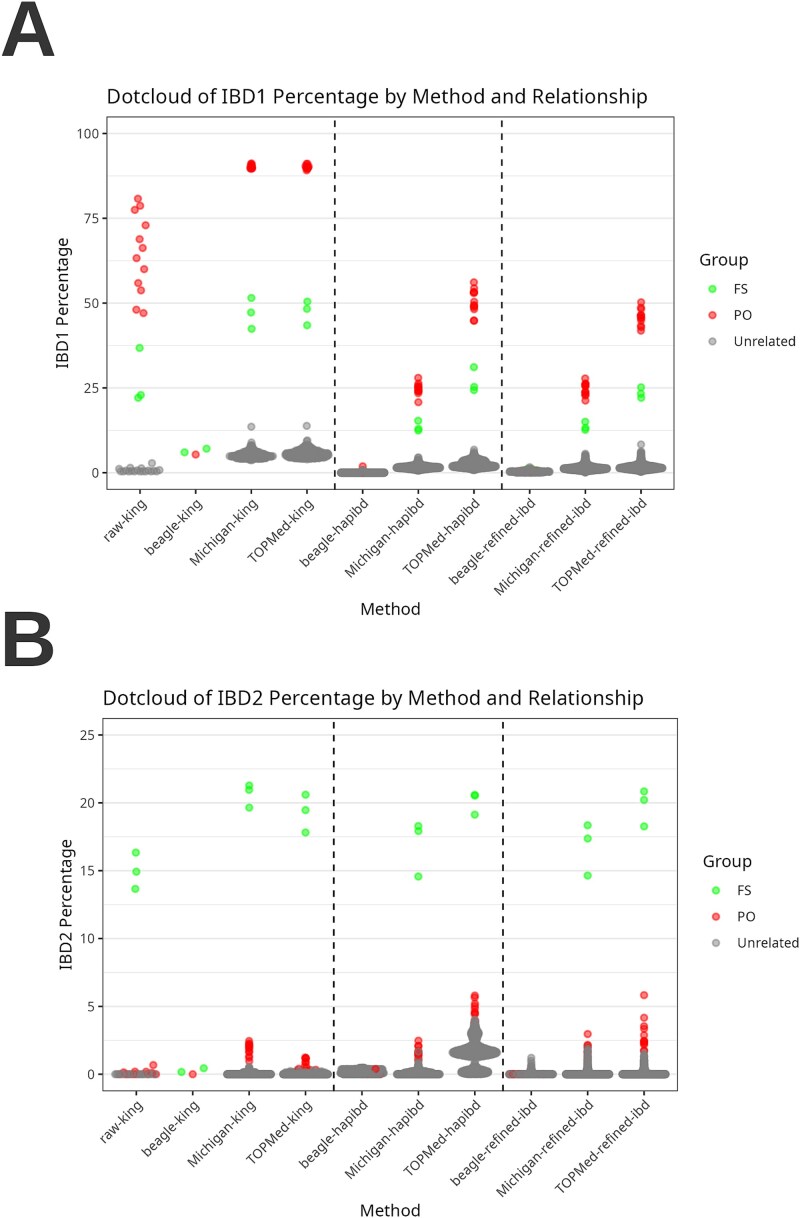
Rare genetic diseases pose significant diagnostic challenges, especially in geographically isolated populations where consanguinity, founder effects, and novel variants often influence disease patterns. Whole-exome sequencing (WES) is standard practice for rare disease diagnostics, but its limited coverage of noncoding regions limits inheritance-by-descent (IBD) and Runs of Homozygosity (RoH) inference. In this study, we tested an imputation-enhanced IBD and RoH detection method using WES data of 84 individuals from 51 families in Boyacá, Colombia-an Andean region with complex admixed American ancestry. By leveraging large, multi-ancestry reference panels to impute genotypes and increase variant distribution, we achieved improved detection of IBD and RoH regions, with KING showing the best results among the different tools that were tested. Imputation with the 1000 Genome reference panel underperformed compared to raw WES data, whereas large reference panels with diverse ancestry showed the best performance. By integrating these refined IBD results with pedigree information, we identified cryptic family relationships, clarified the role of consanguinity, and improved the prioritization of candidate variants. Our findings show that imputation-enhanced IBD analyses can bolster the utility of WES for rare disease studies, contributing to more accurate and timely genetic diagnoses.
期刊介绍:
Human Molecular Genetics concentrates on full-length research papers covering a wide range of topics in all aspects of human molecular genetics. These include:
the molecular basis of human genetic disease
developmental genetics
cancer genetics
neurogenetics
chromosome and genome structure and function
therapy of genetic disease
stem cells in human genetic disease and therapy, including the application of iPS cells
genome-wide association studies
mouse and other models of human diseases
functional genomics
computational genomics
In addition, the journal also publishes research on other model systems for the analysis of genes, especially when there is an obvious relevance to human genetics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


