Zahra Sadeghian, Mohammad Bayat and Davood Gheidari
{"title":"PDK1抑制剂喹唑啉-12- 1衍生物的合成、分子对接、药理评价、MD模拟和DFT计算。","authors":"Zahra Sadeghian, Mohammad Bayat and Davood Gheidari","doi":"10.1039/D5NA00182J","DOIUrl":null,"url":null,"abstract":"<p >3-Phosphoinositide-dependent protein kinase-1(PDK1) is a vital kinase in cellular signaling that regulates growth and survival, playing a crucial role in cancer by activating Akt within the PI3K/Akt pathway. Elevated PDK1 levels correlate with tumor progression and chemotherapy resistance, highlighting its potential as a therapeutic target and biomarker. Schiff bases (SBs) are widely utilized as anticancer agents, as well as for antiviral, antipyretic, antimicrobial, antifungal, antiproliferative, and anti-inflammatory purposes. In this study, we synthesized a series of new quinazolin-12-one derivatives with moderate to good yields (72–92%) and evaluated their efficacy against PDK1 using <em>in silico</em> methods. Comprehensive computational studies, including quantum chemical calculations, molecular docking, molecular dynamics (MD), and absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiling, were performed. Density functional theory (DFT) analysis with the B3LYP/6-31++G (d, p) basis set indicated a promising reactivity profile for the synthesized compounds. The oxygen atoms and π-system of the title compound exhibit high chemical reactivity, serving as electron donor sites and targets for electrophilic attack. Docking analysis with PDK1 enzymes revealed that all compounds, with docking scores between −9.99 and −10.44, demonstrated greater binding affinity than the native ligand, which had a score of −9.49. Among these, compound <strong>3f</strong>, with an energy of −10.44 kcal mol<small><sup>−1</sup></small>, displayed the strongest binding affinity. The MD simulation showed that Ala162 stands out with a notably high interaction fraction, suggesting that it may be a critical residue for the binding affinity of compound <strong>3f</strong>. The analysis of ADMET properties indicated that all inhibitor compounds exhibit favorable pharmacological characteristics, including adherence to Lipinski's Rule of Five (Ro5) as well as the Ghose, Veber, and Egan rules. Additionally, the physicochemical properties demonstrate that all synthesized compounds are capable of human intestinal absorption and have the ability to penetrate the blood–brain barrier (BBB).</p>","PeriodicalId":18806,"journal":{"name":"Nanoscale Advances","volume":" 18","pages":" 5760-5783"},"PeriodicalIF":4.6000,"publicationDate":"2025-08-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12341467/pdf/","citationCount":"0","resultStr":"{\"title\":\"Synthesis, molecular docking, pharmacological evaluation, MD simulation, and DFT calculations of quinazolin-12-one derivatives as PDK1 inhibitors†\",\"authors\":\"Zahra Sadeghian, Mohammad Bayat and Davood Gheidari\",\"doi\":\"10.1039/D5NA00182J\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >3-Phosphoinositide-dependent protein kinase-1(PDK1) is a vital kinase in cellular signaling that regulates growth and survival, playing a crucial role in cancer by activating Akt within the PI3K/Akt pathway. Elevated PDK1 levels correlate with tumor progression and chemotherapy resistance, highlighting its potential as a therapeutic target and biomarker. Schiff bases (SBs) are widely utilized as anticancer agents, as well as for antiviral, antipyretic, antimicrobial, antifungal, antiproliferative, and anti-inflammatory purposes. In this study, we synthesized a series of new quinazolin-12-one derivatives with moderate to good yields (72–92%) and evaluated their efficacy against PDK1 using <em>in silico</em> methods. Comprehensive computational studies, including quantum chemical calculations, molecular docking, molecular dynamics (MD), and absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiling, were performed. Density functional theory (DFT) analysis with the B3LYP/6-31++G (d, p) basis set indicated a promising reactivity profile for the synthesized compounds. The oxygen atoms and π-system of the title compound exhibit high chemical reactivity, serving as electron donor sites and targets for electrophilic attack. Docking analysis with PDK1 enzymes revealed that all compounds, with docking scores between −9.99 and −10.44, demonstrated greater binding affinity than the native ligand, which had a score of −9.49. Among these, compound <strong>3f</strong>, with an energy of −10.44 kcal mol<small><sup>−1</sup></small>, displayed the strongest binding affinity. The MD simulation showed that Ala162 stands out with a notably high interaction fraction, suggesting that it may be a critical residue for the binding affinity of compound <strong>3f</strong>. The analysis of ADMET properties indicated that all inhibitor compounds exhibit favorable pharmacological characteristics, including adherence to Lipinski's Rule of Five (Ro5) as well as the Ghose, Veber, and Egan rules. Additionally, the physicochemical properties demonstrate that all synthesized compounds are capable of human intestinal absorption and have the ability to penetrate the blood–brain barrier (BBB).</p>\",\"PeriodicalId\":18806,\"journal\":{\"name\":\"Nanoscale Advances\",\"volume\":\" 18\",\"pages\":\" 5760-5783\"},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2025-08-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12341467/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nanoscale Advances\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2025/na/d5na00182j\",\"RegionNum\":3,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nanoscale Advances","FirstCategoryId":"88","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/na/d5na00182j","RegionNum":3,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Synthesis, molecular docking, pharmacological evaluation, MD simulation, and DFT calculations of quinazolin-12-one derivatives as PDK1 inhibitors†
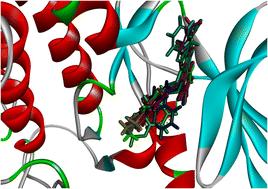
3-Phosphoinositide-dependent protein kinase-1(PDK1) is a vital kinase in cellular signaling that regulates growth and survival, playing a crucial role in cancer by activating Akt within the PI3K/Akt pathway. Elevated PDK1 levels correlate with tumor progression and chemotherapy resistance, highlighting its potential as a therapeutic target and biomarker. Schiff bases (SBs) are widely utilized as anticancer agents, as well as for antiviral, antipyretic, antimicrobial, antifungal, antiproliferative, and anti-inflammatory purposes. In this study, we synthesized a series of new quinazolin-12-one derivatives with moderate to good yields (72–92%) and evaluated their efficacy against PDK1 using in silico methods. Comprehensive computational studies, including quantum chemical calculations, molecular docking, molecular dynamics (MD), and absorption, distribution, metabolism, excretion, and toxicity (ADMET) profiling, were performed. Density functional theory (DFT) analysis with the B3LYP/6-31++G (d, p) basis set indicated a promising reactivity profile for the synthesized compounds. The oxygen atoms and π-system of the title compound exhibit high chemical reactivity, serving as electron donor sites and targets for electrophilic attack. Docking analysis with PDK1 enzymes revealed that all compounds, with docking scores between −9.99 and −10.44, demonstrated greater binding affinity than the native ligand, which had a score of −9.49. Among these, compound 3f, with an energy of −10.44 kcal mol−1, displayed the strongest binding affinity. The MD simulation showed that Ala162 stands out with a notably high interaction fraction, suggesting that it may be a critical residue for the binding affinity of compound 3f. The analysis of ADMET properties indicated that all inhibitor compounds exhibit favorable pharmacological characteristics, including adherence to Lipinski's Rule of Five (Ro5) as well as the Ghose, Veber, and Egan rules. Additionally, the physicochemical properties demonstrate that all synthesized compounds are capable of human intestinal absorption and have the ability to penetrate the blood–brain barrier (BBB).

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


