Jordan Douglas, Remco Bouckaert, Charles W Carter, Peter R Wills
{"title":"减少的氨基酸替代矩阵在现代蛋白质中发现了古代编码字母的痕迹。","authors":"Jordan Douglas, Remco Bouckaert, Charles W Carter, Peter R Wills","doi":"10.1093/molbev/msaf197","DOIUrl":null,"url":null,"abstract":"<p><p>All known living systems make proteins from the same 20 canonically coded amino acids, but this was not always the case. Early genetic coding systems likely operated with a restricted pool of amino acid types and limited means to distinguish between them. Despite this, amino acid substitution models like LG and WAG all assume a constant coding alphabet over time. That makes them especially inappropriate for the aminoacyl-tRNA synthetases (aaRS)-the enzymes that govern translation. To address this limitation, we created a class of substitution models that account for evolutionary changes in the coding alphabet size by defining the transition from 19 states in a past epoch to 20 now. We use a Bayesian phylogenetic framework to improve phylogeny estimation and testing of this two-alphabet hypothesis. The hypothesis was strongly rejected by datasets composed exclusively of \"young\" eukaryotic proteins. It was generally supported by \"old\" (aaRS and non-aaRS) proteins whose origins date from before the last universal common ancestor. Standard methods overestimate the divergence ages of proteins that originated under reduced coding alphabets in both simulated and aaRS alignments. The new model provides a timeline slightly more consistent with the Earth's history. Our findings suggest that aaRS functional bifurcation events can explain much of the genetic code's evolution, but there remain other unknown forces at play too. This work provides a robust, seamless framework for reconstructing phylogenies from ancient protein datasets and offers further insights into the dawn of molecular biology.</p>","PeriodicalId":18730,"journal":{"name":"Molecular biology and evolution","volume":" ","pages":""},"PeriodicalIF":5.3000,"publicationDate":"2025-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12402984/pdf/","citationCount":"0","resultStr":"{\"title\":\"Reduced Amino Acid Substitution Matrices Find Traces of Ancient Coding Alphabets in Modern Day Proteins.\",\"authors\":\"Jordan Douglas, Remco Bouckaert, Charles W Carter, Peter R Wills\",\"doi\":\"10.1093/molbev/msaf197\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>All known living systems make proteins from the same 20 canonically coded amino acids, but this was not always the case. Early genetic coding systems likely operated with a restricted pool of amino acid types and limited means to distinguish between them. Despite this, amino acid substitution models like LG and WAG all assume a constant coding alphabet over time. That makes them especially inappropriate for the aminoacyl-tRNA synthetases (aaRS)-the enzymes that govern translation. To address this limitation, we created a class of substitution models that account for evolutionary changes in the coding alphabet size by defining the transition from 19 states in a past epoch to 20 now. We use a Bayesian phylogenetic framework to improve phylogeny estimation and testing of this two-alphabet hypothesis. The hypothesis was strongly rejected by datasets composed exclusively of \\\"young\\\" eukaryotic proteins. It was generally supported by \\\"old\\\" (aaRS and non-aaRS) proteins whose origins date from before the last universal common ancestor. Standard methods overestimate the divergence ages of proteins that originated under reduced coding alphabets in both simulated and aaRS alignments. The new model provides a timeline slightly more consistent with the Earth's history. Our findings suggest that aaRS functional bifurcation events can explain much of the genetic code's evolution, but there remain other unknown forces at play too. This work provides a robust, seamless framework for reconstructing phylogenies from ancient protein datasets and offers further insights into the dawn of molecular biology.</p>\",\"PeriodicalId\":18730,\"journal\":{\"name\":\"Molecular biology and evolution\",\"volume\":\" \",\"pages\":\"\"},\"PeriodicalIF\":5.3000,\"publicationDate\":\"2025-09-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12402984/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular biology and evolution\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/molbev/msaf197\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular biology and evolution","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/molbev/msaf197","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Reduced Amino Acid Substitution Matrices Find Traces of Ancient Coding Alphabets in Modern Day Proteins.
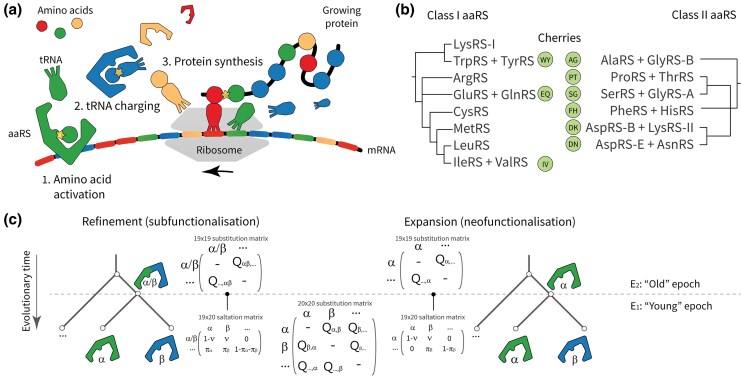
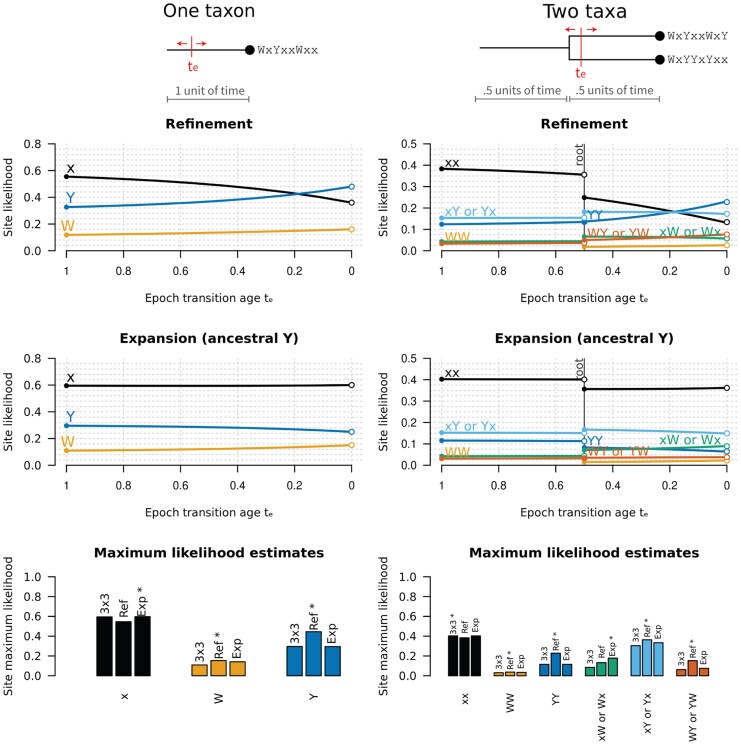
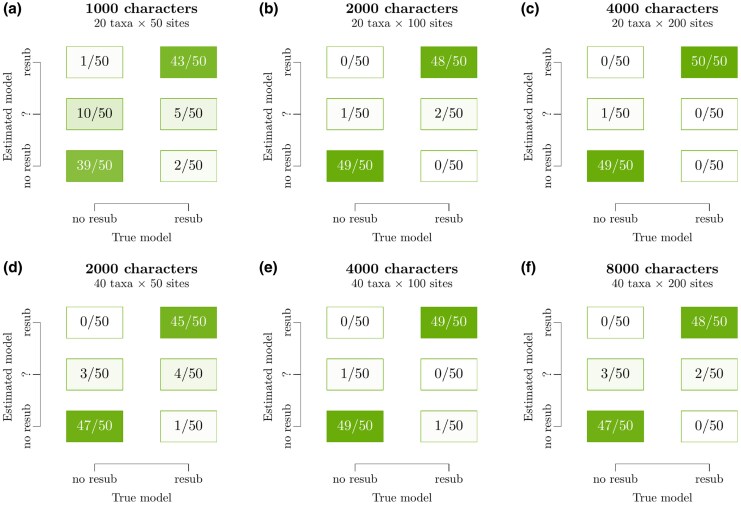
All known living systems make proteins from the same 20 canonically coded amino acids, but this was not always the case. Early genetic coding systems likely operated with a restricted pool of amino acid types and limited means to distinguish between them. Despite this, amino acid substitution models like LG and WAG all assume a constant coding alphabet over time. That makes them especially inappropriate for the aminoacyl-tRNA synthetases (aaRS)-the enzymes that govern translation. To address this limitation, we created a class of substitution models that account for evolutionary changes in the coding alphabet size by defining the transition from 19 states in a past epoch to 20 now. We use a Bayesian phylogenetic framework to improve phylogeny estimation and testing of this two-alphabet hypothesis. The hypothesis was strongly rejected by datasets composed exclusively of "young" eukaryotic proteins. It was generally supported by "old" (aaRS and non-aaRS) proteins whose origins date from before the last universal common ancestor. Standard methods overestimate the divergence ages of proteins that originated under reduced coding alphabets in both simulated and aaRS alignments. The new model provides a timeline slightly more consistent with the Earth's history. Our findings suggest that aaRS functional bifurcation events can explain much of the genetic code's evolution, but there remain other unknown forces at play too. This work provides a robust, seamless framework for reconstructing phylogenies from ancient protein datasets and offers further insights into the dawn of molecular biology.
期刊介绍:
Molecular Biology and Evolution
Journal Overview:
Publishes research at the interface of molecular (including genomics) and evolutionary biology
Considers manuscripts containing patterns, processes, and predictions at all levels of organization: population, taxonomic, functional, and phenotypic
Interested in fundamental discoveries, new and improved methods, resources, technologies, and theories advancing evolutionary research
Publishes balanced reviews of recent developments in genome evolution and forward-looking perspectives suggesting future directions in molecular evolution applications.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


