l达克- kvik执行快速和强大的混合模型关联分析定量和二元表型
IF 29
1区 生物学
Q1 GENETICS & HEREDITY
引用次数: 0
摘要
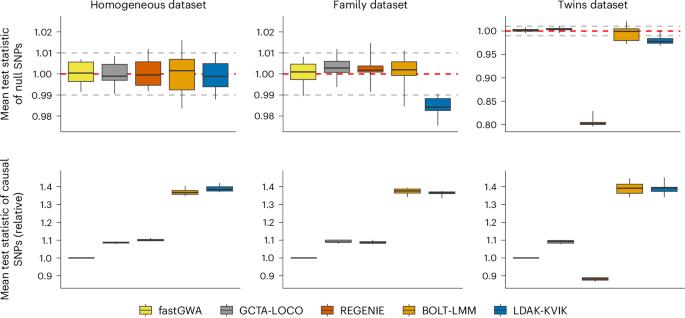
混合模型关联分析(MMAA)是进行全基因组关联研究的首选工具。但是,现有的MMAA工具通常具有较长的运行时间和较高的内存需求。在这里,我们提出l达克- kvik,一个MMAA工具,用于分析定量和二元表型。ltak - kvik的计算效率很高,只需不到10个CPU小时和5gb内存就可以分析35万人的全基因组数据。通过模拟表型,我们发现ltak - kvik可以为同质和异质数据集提供校准良好的测试统计。当应用于真实表型时,l达克- kvik在所有考虑的工具中具有最高的功效。例如,在40个定量UK Biobank表型(平均样样量为349,000)中,ltak - kvik发现的独立的全基因组显著位点比经典线性回归多16%,而BOLT-LMM和REGENIE分别发现了15%和11%。l达克- kvik也可用于进行基于基因的检测;在40个定量的英国生物银行表型中,LDAK-KVIK比现有的主要工具多发现18%的重要基因。最后,LDAK-KVIK产生最先进的多基因评分。本文章由计算机程序翻译,如有差异,请以英文原文为准。
LDAK-KVIK performs fast and powerful mixed-model association analysis of quantitative and binary phenotypes
Mixed-model association analysis (MMAA) is the preferred tool for performing genome-wide association studies. However, existing MMAA tools often have long runtimes and high memory requirements. Here we present LDAK-KVIK, an MMAA tool for analysis of quantitative and binary phenotypes. LDAK-KVIK is computationally efficient, requiring less than 10 CPU hours and 5 Gb memory to analyze genome-wide data for 350,000 individuals. Using simulated phenotypes, we show that LDAK-KVIK produces well-calibrated test statistics for both homogeneous and heterogeneous datasets. When applied to real phenotypes, LDAK-KVIK has the highest power among all tools considered. For example, across 40 quantitative UK Biobank phenotypes (average sample size 349,000), LDAK-KVIK finds 16% more independent, genome-wide significant loci than classical linear regression, whereas BOLT-LMM and REGENIE find 15% and 11% more, respectively. LDAK-KVIK can also be used to perform gene-based tests; across the 40 quantitative UK Biobank phenotypes, LDAK-KVIK finds 18% more significant genes than the leading existing tool. Last, LDAK-KVIK produces state-of-the-art polygenic scores. LDAK-KVIK is a mixed-model association method for genome-wide studies that optimizes computational performance and power. LDAK-KVIK can also perform gene-based tests and produces state-of-the-art polygenic scores.
求助全文
通过发布文献求助,成功后即可免费获取论文全文。
去求助
来源期刊

Nature genetics
生物-遗传学
CiteScore
43.00
自引率
2.60%
发文量
241
审稿时长
3 months
期刊介绍:
Nature Genetics publishes the very highest quality research in genetics. It encompasses genetic and functional genomic studies on human and plant traits and on other model organisms. Current emphasis is on the genetic basis for common and complex diseases and on the functional mechanism, architecture and evolution of gene networks, studied by experimental perturbation.
Integrative genetic topics comprise, but are not limited to:
-Genes in the pathology of human disease
-Molecular analysis of simple and complex genetic traits
-Cancer genetics
-Agricultural genomics
-Developmental genetics
-Regulatory variation in gene expression
-Strategies and technologies for extracting function from genomic data
-Pharmacological genomics
-Genome evolution
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


