Olivier Jeannin, Eun-Hye Jang, Marc Fourmigué*, Enrique Espinosa and Emmanuel Aubert*,
{"title":"晶体环境对I-Cl加合物中卤键对取代吡啶的影响","authors":"Olivier Jeannin, Eun-Hye Jang, Marc Fourmigué*, Enrique Espinosa and Emmanuel Aubert*, ","doi":"10.1021/acs.cgd.5c00646","DOIUrl":null,"url":null,"abstract":"<p >Strong halogen bond (HaB) interactions between six different <i>para</i>-substituted pyridines [R–Py] and I–Cl (R = CF<sub>3</sub>, CO<sub>2</sub>Me, H, Me, NMe<sub>2</sub>, and pyrrolidin-1-yl) lead to the formation of [R–Py]•I–Cl adducts, which can be considered as a unique molecular entity, as deduced from both energetic and topological analyses, with the strongest HaB observed with the most electron-rich pyridines. All six adducts at optimized geometries show HaB interactions weaker than those found at experimental geometries, highlighting the effect of the crystalline environment on such HaB systems. Indeed, when neighboring adducts associated through secondary C–H···Cl interactions are explicitly considered in the geometry optimizations, the experimental N···I···Cl geometries are then convincingly reproduced, paralleling the effect of an implicit PCM solvent model. The effects of such weaker secondary hydrogen bond interactions are also well reproduced by considering one single [Py]•I–Cl adduct interacting with HF molecules through weak F–H···Cl hydrogen bonds along the series [Py]•ICl < [Py]•ICl•HF < [Py]•ICl•2HF. The topological properties at both N<sub>Py</sub>···I/I···Cl bond critical points follow the evolving behavior of the concomitant increase/decrease of both interaction strengths while paralleling the shortening/lengthening of their internuclear distances. Furthermore, the analysis of the topological Cl and I atomic net charges, as well as the dipole moment of the adducts, points out the increase of charge separation [N–I<sup>δ</sup><sup>+</sup>–Cl<sup>δ</sup><sup>–</sup>] within the adduct with that of the N<sub>Py</sub>···I interaction strength and its concomitant shortening. Complementary ELF and Feynman force calculations (to determine the ionic vs. covalent vs. charge-shift components) show that the halogen-bonded [R-Py]•ICl adducts switch from a closed-shell interaction with an incipient covalent character to a dative covalent bond, when considering explicitly the molecular crystalline environment.</p>","PeriodicalId":34,"journal":{"name":"Crystal Growth & Design","volume":"25 15","pages":"6204–6213"},"PeriodicalIF":3.4000,"publicationDate":"2025-07-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Crystal Environment Effects on Halogen-Bonded para-Substituted Pyridines in Their I–Cl Adducts\",\"authors\":\"Olivier Jeannin, Eun-Hye Jang, Marc Fourmigué*, Enrique Espinosa and Emmanuel Aubert*, \",\"doi\":\"10.1021/acs.cgd.5c00646\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >Strong halogen bond (HaB) interactions between six different <i>para</i>-substituted pyridines [R–Py] and I–Cl (R = CF<sub>3</sub>, CO<sub>2</sub>Me, H, Me, NMe<sub>2</sub>, and pyrrolidin-1-yl) lead to the formation of [R–Py]•I–Cl adducts, which can be considered as a unique molecular entity, as deduced from both energetic and topological analyses, with the strongest HaB observed with the most electron-rich pyridines. All six adducts at optimized geometries show HaB interactions weaker than those found at experimental geometries, highlighting the effect of the crystalline environment on such HaB systems. Indeed, when neighboring adducts associated through secondary C–H···Cl interactions are explicitly considered in the geometry optimizations, the experimental N···I···Cl geometries are then convincingly reproduced, paralleling the effect of an implicit PCM solvent model. The effects of such weaker secondary hydrogen bond interactions are also well reproduced by considering one single [Py]•I–Cl adduct interacting with HF molecules through weak F–H···Cl hydrogen bonds along the series [Py]•ICl < [Py]•ICl•HF < [Py]•ICl•2HF. The topological properties at both N<sub>Py</sub>···I/I···Cl bond critical points follow the evolving behavior of the concomitant increase/decrease of both interaction strengths while paralleling the shortening/lengthening of their internuclear distances. Furthermore, the analysis of the topological Cl and I atomic net charges, as well as the dipole moment of the adducts, points out the increase of charge separation [N–I<sup>δ</sup><sup>+</sup>–Cl<sup>δ</sup><sup>–</sup>] within the adduct with that of the N<sub>Py</sub>···I interaction strength and its concomitant shortening. Complementary ELF and Feynman force calculations (to determine the ionic vs. covalent vs. charge-shift components) show that the halogen-bonded [R-Py]•ICl adducts switch from a closed-shell interaction with an incipient covalent character to a dative covalent bond, when considering explicitly the molecular crystalline environment.</p>\",\"PeriodicalId\":34,\"journal\":{\"name\":\"Crystal Growth & Design\",\"volume\":\"25 15\",\"pages\":\"6204–6213\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2025-07-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Crystal Growth & Design\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.acs.org/doi/10.1021/acs.cgd.5c00646\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Crystal Growth & Design","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.cgd.5c00646","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
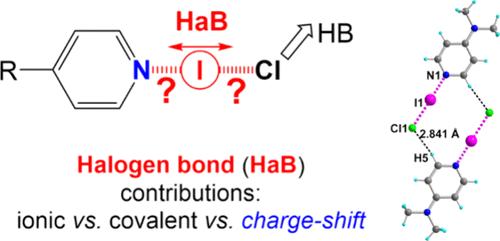
Crystal Environment Effects on Halogen-Bonded para-Substituted Pyridines in Their I–Cl Adducts
Strong halogen bond (HaB) interactions between six different para-substituted pyridines [R–Py] and I–Cl (R = CF3, CO2Me, H, Me, NMe2, and pyrrolidin-1-yl) lead to the formation of [R–Py]•I–Cl adducts, which can be considered as a unique molecular entity, as deduced from both energetic and topological analyses, with the strongest HaB observed with the most electron-rich pyridines. All six adducts at optimized geometries show HaB interactions weaker than those found at experimental geometries, highlighting the effect of the crystalline environment on such HaB systems. Indeed, when neighboring adducts associated through secondary C–H···Cl interactions are explicitly considered in the geometry optimizations, the experimental N···I···Cl geometries are then convincingly reproduced, paralleling the effect of an implicit PCM solvent model. The effects of such weaker secondary hydrogen bond interactions are also well reproduced by considering one single [Py]•I–Cl adduct interacting with HF molecules through weak F–H···Cl hydrogen bonds along the series [Py]•ICl < [Py]•ICl•HF < [Py]•ICl•2HF. The topological properties at both NPy···I/I···Cl bond critical points follow the evolving behavior of the concomitant increase/decrease of both interaction strengths while paralleling the shortening/lengthening of their internuclear distances. Furthermore, the analysis of the topological Cl and I atomic net charges, as well as the dipole moment of the adducts, points out the increase of charge separation [N–Iδ+–Clδ–] within the adduct with that of the NPy···I interaction strength and its concomitant shortening. Complementary ELF and Feynman force calculations (to determine the ionic vs. covalent vs. charge-shift components) show that the halogen-bonded [R-Py]•ICl adducts switch from a closed-shell interaction with an incipient covalent character to a dative covalent bond, when considering explicitly the molecular crystalline environment.
期刊介绍:
The aim of Crystal Growth & Design is to stimulate crossfertilization of knowledge among scientists and engineers working in the fields of crystal growth, crystal engineering, and the industrial application of crystalline materials.
Crystal Growth & Design publishes theoretical and experimental studies of the physical, chemical, and biological phenomena and processes related to the design, growth, and application of crystalline materials. Synergistic approaches originating from different disciplines and technologies and integrating the fields of crystal growth, crystal engineering, intermolecular interactions, and industrial application are encouraged.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


