Jean Solarz, Christelle Soukaseum, Stéphane Frémont, Sébastien Eymieux, Camilia Nabli, Christelle Repérant, Elisa Rossi, Jean-Claude Bordet, Cécile V. Denis, Pierre Mangin, Yacine Boulaftali, R. Jeroen Pasterkamp, Hana Raslova, Dominique Baruch, Frédéric Adam, Arnaud Echard, Alexandre Kauskot
{"title":"氧化还原酶MICAL1分解f -肌动蛋白促进血小板中机械依赖性VWF-GPIbα相互作用","authors":"Jean Solarz, Christelle Soukaseum, Stéphane Frémont, Sébastien Eymieux, Camilia Nabli, Christelle Repérant, Elisa Rossi, Jean-Claude Bordet, Cécile V. Denis, Pierre Mangin, Yacine Boulaftali, R. Jeroen Pasterkamp, Hana Raslova, Dominique Baruch, Frédéric Adam, Arnaud Echard, Alexandre Kauskot","doi":"10.1038/s41467-025-62487-2","DOIUrl":null,"url":null,"abstract":"<p>Mechano-dependent interactions are key to thrombus formation and hemostasis, enabling stable platelet adhesion to injured vessels. The interaction between von Willebrand factor (VWF) and the platelet receptor GPIb-IX-V is central to this process. While GPIbα connects to the actin cytoskeleton, whether actin dynamics are important for GPIbα function under hemodynamic, high shear conditions remains largely unknown. Here, we show that actin disassembly is critical for proper VWF-GPIbα binding under shear. Mechanistically, we identify the oxidoreductase MICAL1 as a shear-activated regulator that promotes local F-actin disassembly around the GPIb-IX-V complex. This enables its translocation to lipid rafts and reinforces VWF binding. MICAL1-deficient platelets display impaired adhesion, increased deformability under shear, and defective thrombus formation in vivo. Thus, MICAL1 drives shear-dependent actin remodeling that supports GPIb-IX-V mechanotransduction and platelet function. These findings uncover a role for actin oxidation in platelet adhesion, providing a connection between cytoskeletal redox control and platelet function during thrombus formation.</p>","PeriodicalId":19066,"journal":{"name":"Nature Communications","volume":"1 1","pages":""},"PeriodicalIF":15.7000,"publicationDate":"2025-08-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"F-actin disassembly by the oxidoreductase MICAL1 promotes mechano-dependent VWF-GPIbα interaction in platelets\",\"authors\":\"Jean Solarz, Christelle Soukaseum, Stéphane Frémont, Sébastien Eymieux, Camilia Nabli, Christelle Repérant, Elisa Rossi, Jean-Claude Bordet, Cécile V. Denis, Pierre Mangin, Yacine Boulaftali, R. Jeroen Pasterkamp, Hana Raslova, Dominique Baruch, Frédéric Adam, Arnaud Echard, Alexandre Kauskot\",\"doi\":\"10.1038/s41467-025-62487-2\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Mechano-dependent interactions are key to thrombus formation and hemostasis, enabling stable platelet adhesion to injured vessels. The interaction between von Willebrand factor (VWF) and the platelet receptor GPIb-IX-V is central to this process. While GPIbα connects to the actin cytoskeleton, whether actin dynamics are important for GPIbα function under hemodynamic, high shear conditions remains largely unknown. Here, we show that actin disassembly is critical for proper VWF-GPIbα binding under shear. Mechanistically, we identify the oxidoreductase MICAL1 as a shear-activated regulator that promotes local F-actin disassembly around the GPIb-IX-V complex. This enables its translocation to lipid rafts and reinforces VWF binding. MICAL1-deficient platelets display impaired adhesion, increased deformability under shear, and defective thrombus formation in vivo. Thus, MICAL1 drives shear-dependent actin remodeling that supports GPIb-IX-V mechanotransduction and platelet function. These findings uncover a role for actin oxidation in platelet adhesion, providing a connection between cytoskeletal redox control and platelet function during thrombus formation.</p>\",\"PeriodicalId\":19066,\"journal\":{\"name\":\"Nature Communications\",\"volume\":\"1 1\",\"pages\":\"\"},\"PeriodicalIF\":15.7000,\"publicationDate\":\"2025-08-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature Communications\",\"FirstCategoryId\":\"103\",\"ListUrlMain\":\"https://doi.org/10.1038/s41467-025-62487-2\",\"RegionNum\":1,\"RegionCategory\":\"综合性期刊\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MULTIDISCIPLINARY SCIENCES\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Communications","FirstCategoryId":"103","ListUrlMain":"https://doi.org/10.1038/s41467-025-62487-2","RegionNum":1,"RegionCategory":"综合性期刊","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MULTIDISCIPLINARY SCIENCES","Score":null,"Total":0}
F-actin disassembly by the oxidoreductase MICAL1 promotes mechano-dependent VWF-GPIbα interaction in platelets
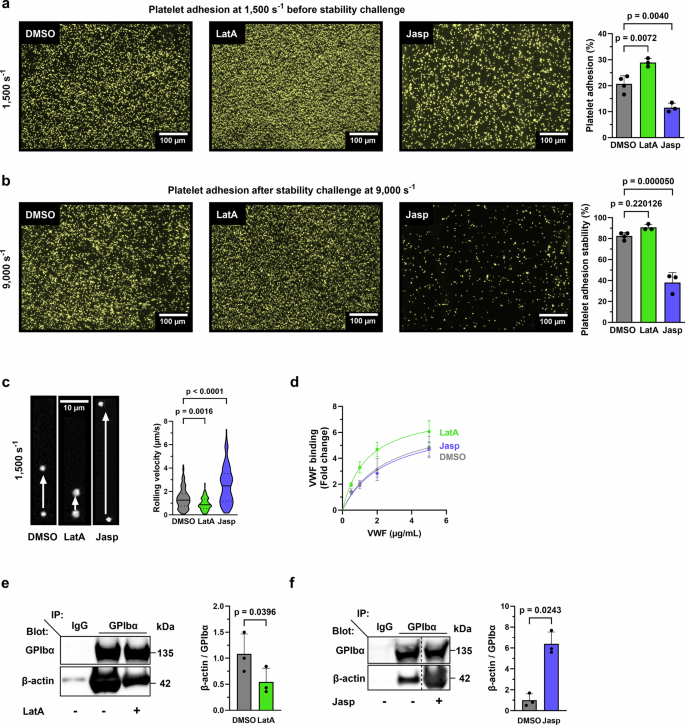
Mechano-dependent interactions are key to thrombus formation and hemostasis, enabling stable platelet adhesion to injured vessels. The interaction between von Willebrand factor (VWF) and the platelet receptor GPIb-IX-V is central to this process. While GPIbα connects to the actin cytoskeleton, whether actin dynamics are important for GPIbα function under hemodynamic, high shear conditions remains largely unknown. Here, we show that actin disassembly is critical for proper VWF-GPIbα binding under shear. Mechanistically, we identify the oxidoreductase MICAL1 as a shear-activated regulator that promotes local F-actin disassembly around the GPIb-IX-V complex. This enables its translocation to lipid rafts and reinforces VWF binding. MICAL1-deficient platelets display impaired adhesion, increased deformability under shear, and defective thrombus formation in vivo. Thus, MICAL1 drives shear-dependent actin remodeling that supports GPIb-IX-V mechanotransduction and platelet function. These findings uncover a role for actin oxidation in platelet adhesion, providing a connection between cytoskeletal redox control and platelet function during thrombus formation.
期刊介绍:
Nature Communications, an open-access journal, publishes high-quality research spanning all areas of the natural sciences. Papers featured in the journal showcase significant advances relevant to specialists in each respective field. With a 2-year impact factor of 16.6 (2022) and a median time of 8 days from submission to the first editorial decision, Nature Communications is committed to rapid dissemination of research findings. As a multidisciplinary journal, it welcomes contributions from biological, health, physical, chemical, Earth, social, mathematical, applied, and engineering sciences, aiming to highlight important breakthroughs within each domain.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


