Hanan AlQudairy, Mohammad A AlMuhaizea, Mohamed Tohary, Maissa Alfuraih, Aisha Alnafisah, Aljouhra AlHargan, Anoud Albader, Hadeel Jaber, Rawan Almass, Albandary Albakheet, Terfa Alsheddi, Eman AlObeid, Maha M Alrasheed, Ali Al-Odaib, Hamad AlZaidan, Moeenaldeen D AlSayed, Namik Kaya
{"title":"sptbn4相关神经发育障碍伴张力低下、神经病变和耳聋的自然病史","authors":"Hanan AlQudairy, Mohammad A AlMuhaizea, Mohamed Tohary, Maissa Alfuraih, Aisha Alnafisah, Aljouhra AlHargan, Anoud Albader, Hadeel Jaber, Rawan Almass, Albandary Albakheet, Terfa Alsheddi, Eman AlObeid, Maha M Alrasheed, Ali Al-Odaib, Hamad AlZaidan, Moeenaldeen D AlSayed, Namik Kaya","doi":"10.1186/s13023-025-03810-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Pathogenic variants in SPTBN4 have been linked to autosomal recessive \"neurodevelopmental disorder with hypotonia, neuropathy, and deafness\" (MIM# 617519) known as NEDHND. The disorder is highlighted with neuropathy, muscle weakness, and infrequent appearance of seizures in the affected individuals. This study aims to investigate the natural history of the disease, present genetic and clinical appearance of the syndrome in a highly consanguineous population, Saudi Arabia, and finally provide an overview of the reported cases, their clinical features, and disease-causing variants.</p><p><strong>Methods: </strong>The study started with a search through neurology clinics and local databases and utilized genetic testing records after diagnosing a patient with NEDHND at our hospital (King Faisal Specialist Hospital and Research Centre, KFSHRC). Based on the search we have identified additional patients (in total, n = 10) with the disease and performed genetic testing using whole exome sequencing and confirmatory Sanger sequencing. We performed RT-PCR on RNA extracted from lymphoblastoid cell line from a patient who found to have an aberrant splicing variant. Finally, we comprehensively reviewed current literature and available data related to the disease.</p><p><strong>Results: </strong>We present natural history of SPTBN4-associated neurodevelopmental disorder with hypotonia, neuropathy, and deafness in addition to four Saudi families with ten affected individuals who share clinical features of NEDHND. We report three known mutations and one novel nonsense variant, highlight atypical clinical features related to cerebellar involvement, confirm the pathogenicity of a splicing variant by RT-PCR, and review the findings of previously reported patients.</p><p><strong>Conclusion: </strong>Our study defines the clinical phenotype of a cohort of NEDHND in detail including the evolution of patients' clinical features, compares them to previously reported cases, and utilizes the existing data on the disease to direct development of a better prevention plan by means of genetic and preimplantation counseling. Our study may help and enable future clinical trials focusing on NEDHND in our country.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"415"},"PeriodicalIF":3.5000,"publicationDate":"2025-08-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12335179/pdf/","citationCount":"0","resultStr":"{\"title\":\"Natural history of SPTBN4-related neurodevelopmental disorder with hypotonia, neuropathy, and deafness.\",\"authors\":\"Hanan AlQudairy, Mohammad A AlMuhaizea, Mohamed Tohary, Maissa Alfuraih, Aisha Alnafisah, Aljouhra AlHargan, Anoud Albader, Hadeel Jaber, Rawan Almass, Albandary Albakheet, Terfa Alsheddi, Eman AlObeid, Maha M Alrasheed, Ali Al-Odaib, Hamad AlZaidan, Moeenaldeen D AlSayed, Namik Kaya\",\"doi\":\"10.1186/s13023-025-03810-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Pathogenic variants in SPTBN4 have been linked to autosomal recessive \\\"neurodevelopmental disorder with hypotonia, neuropathy, and deafness\\\" (MIM# 617519) known as NEDHND. The disorder is highlighted with neuropathy, muscle weakness, and infrequent appearance of seizures in the affected individuals. This study aims to investigate the natural history of the disease, present genetic and clinical appearance of the syndrome in a highly consanguineous population, Saudi Arabia, and finally provide an overview of the reported cases, their clinical features, and disease-causing variants.</p><p><strong>Methods: </strong>The study started with a search through neurology clinics and local databases and utilized genetic testing records after diagnosing a patient with NEDHND at our hospital (King Faisal Specialist Hospital and Research Centre, KFSHRC). Based on the search we have identified additional patients (in total, n = 10) with the disease and performed genetic testing using whole exome sequencing and confirmatory Sanger sequencing. We performed RT-PCR on RNA extracted from lymphoblastoid cell line from a patient who found to have an aberrant splicing variant. Finally, we comprehensively reviewed current literature and available data related to the disease.</p><p><strong>Results: </strong>We present natural history of SPTBN4-associated neurodevelopmental disorder with hypotonia, neuropathy, and deafness in addition to four Saudi families with ten affected individuals who share clinical features of NEDHND. We report three known mutations and one novel nonsense variant, highlight atypical clinical features related to cerebellar involvement, confirm the pathogenicity of a splicing variant by RT-PCR, and review the findings of previously reported patients.</p><p><strong>Conclusion: </strong>Our study defines the clinical phenotype of a cohort of NEDHND in detail including the evolution of patients' clinical features, compares them to previously reported cases, and utilizes the existing data on the disease to direct development of a better prevention plan by means of genetic and preimplantation counseling. Our study may help and enable future clinical trials focusing on NEDHND in our country.</p>\",\"PeriodicalId\":19651,\"journal\":{\"name\":\"Orphanet Journal of Rare Diseases\",\"volume\":\"20 1\",\"pages\":\"415\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2025-08-08\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12335179/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Orphanet Journal of Rare Diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13023-025-03810-4\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-03810-4","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Natural history of SPTBN4-related neurodevelopmental disorder with hypotonia, neuropathy, and deafness.
Background: Pathogenic variants in SPTBN4 have been linked to autosomal recessive "neurodevelopmental disorder with hypotonia, neuropathy, and deafness" (MIM# 617519) known as NEDHND. The disorder is highlighted with neuropathy, muscle weakness, and infrequent appearance of seizures in the affected individuals. This study aims to investigate the natural history of the disease, present genetic and clinical appearance of the syndrome in a highly consanguineous population, Saudi Arabia, and finally provide an overview of the reported cases, their clinical features, and disease-causing variants.
Methods: The study started with a search through neurology clinics and local databases and utilized genetic testing records after diagnosing a patient with NEDHND at our hospital (King Faisal Specialist Hospital and Research Centre, KFSHRC). Based on the search we have identified additional patients (in total, n = 10) with the disease and performed genetic testing using whole exome sequencing and confirmatory Sanger sequencing. We performed RT-PCR on RNA extracted from lymphoblastoid cell line from a patient who found to have an aberrant splicing variant. Finally, we comprehensively reviewed current literature and available data related to the disease.
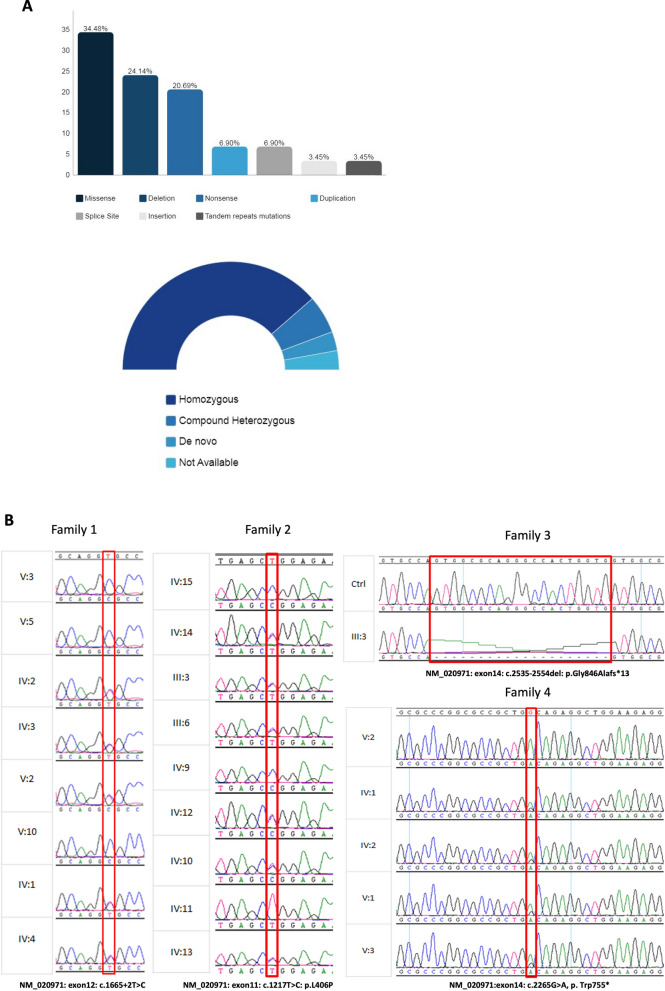
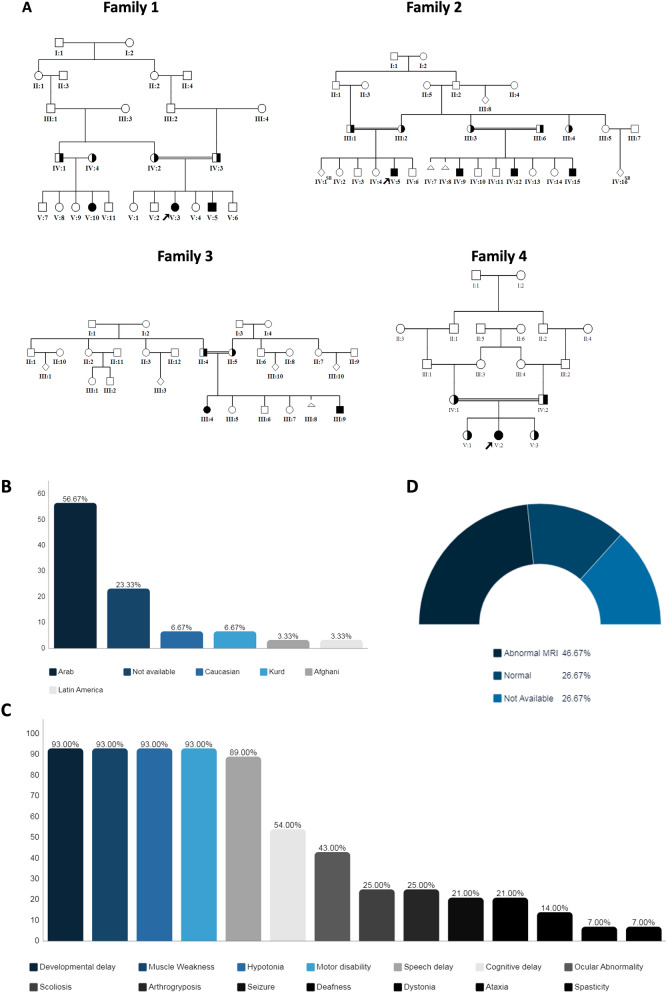
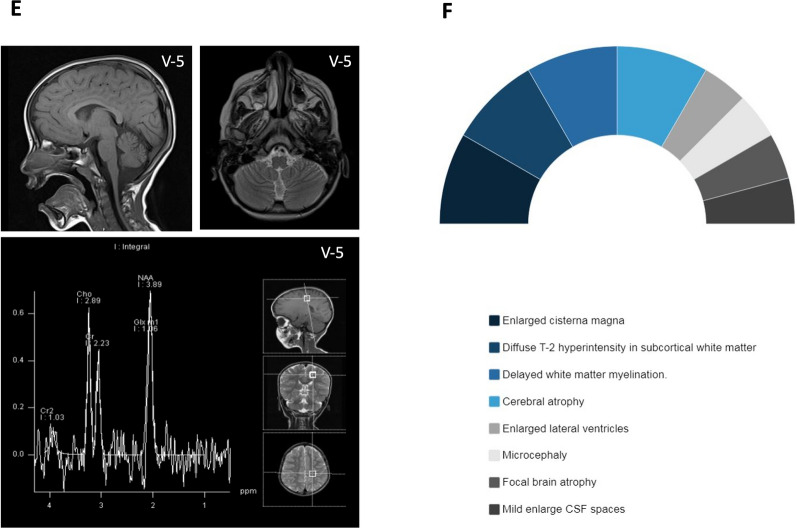
Results: We present natural history of SPTBN4-associated neurodevelopmental disorder with hypotonia, neuropathy, and deafness in addition to four Saudi families with ten affected individuals who share clinical features of NEDHND. We report three known mutations and one novel nonsense variant, highlight atypical clinical features related to cerebellar involvement, confirm the pathogenicity of a splicing variant by RT-PCR, and review the findings of previously reported patients.
Conclusion: Our study defines the clinical phenotype of a cohort of NEDHND in detail including the evolution of patients' clinical features, compares them to previously reported cases, and utilizes the existing data on the disease to direct development of a better prevention plan by means of genetic and preimplantation counseling. Our study may help and enable future clinical trials focusing on NEDHND in our country.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


