{"title":"肿瘤葡萄糖重编程抑制铜增生:综述。","authors":"Xiao-Hang Song, Yi-Hang Ding, Jing-Song Chen","doi":"10.17305/bb.2025.12751","DOIUrl":null,"url":null,"abstract":"<p><p>Cuproptosis is a copper-dependent form of regulated cell death that begins when ferredoxin 1 (FDX1) reduces Cu²⁺ to Cu¹⁺, allowing the ion to bind lipoylated enzymes of the tricarboxylic-acid (TCA) cycle, drive protein aggregation, dismantle iron-sulphur clusters and trigger fatal proteotoxic stress. Most tumours, despite accumulating copper, evade this fate through glucose-metabolic rewiring. First, oncogenic stabilisation of hypoxia-inducible factor-1 alpha (HIF-1α) and MYC increases pyruvate dehydrogenase kinase (PDK) activity, which phosphorylates and inactivates the pyruvate dehydrogenase complex (PDC), shrinking the lipoylated target pool in mitochondria and cutting the feed into the TCA cycle. Second, glycolytic signalling suppresses cuproptosis-promoting genes such as FDX1 and dihydrolipoamide S-acetyltransferase while inducing the negative regulator glutaminase (GLS), further lowering copper sensitivity. Third, diversion of glycolytic intermediates into the pentose-phosphate pathway (PPP) supplies abundant nicotinamide adenine dinucleotide phosphate (NADPH), whereas enhanced glutamine catabolism furnishes glutamate; together these fuels expand reduced glutathione (GSH) and metallothionein (MT) pools that chelate Cu¹⁺ and quench reactive oxygen species exactly where cuproptosis is executed. Consequently, glycolysis-dependent cancer cells are far less sensitive to copper-ionophore drugs such as elesclomol or disulfiram than respiration-dependent counterparts, and clinical datasets consistently link high PDK and low PDC-subunit expression with poor prognosis. These insights highlight rational combination strategies: re-activating the TCA cycle with PDK inhibitors, draining PPP- or GLS-driven NADPH/GSH supply, and concurrently delivering copper ionophores could reopen the cuproptotic trap in tumours. Validating such approaches in vivo, charting upstream regulators of FDX1 and mapping crosstalk between cuproptosis and other lethal programmes remain key steps toward exploiting this copper-centred vulnerability in cancer therapy.</p>","PeriodicalId":72398,"journal":{"name":"Biomolecules & biomedicine","volume":" ","pages":"251-261"},"PeriodicalIF":0.0000,"publicationDate":"2025-08-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12505534/pdf/","citationCount":"0","resultStr":"{\"title\":\"Tumor glucose reprogramming suppresses cuproptosis: A review.\",\"authors\":\"Xiao-Hang Song, Yi-Hang Ding, Jing-Song Chen\",\"doi\":\"10.17305/bb.2025.12751\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Cuproptosis is a copper-dependent form of regulated cell death that begins when ferredoxin 1 (FDX1) reduces Cu²⁺ to Cu¹⁺, allowing the ion to bind lipoylated enzymes of the tricarboxylic-acid (TCA) cycle, drive protein aggregation, dismantle iron-sulphur clusters and trigger fatal proteotoxic stress. Most tumours, despite accumulating copper, evade this fate through glucose-metabolic rewiring. First, oncogenic stabilisation of hypoxia-inducible factor-1 alpha (HIF-1α) and MYC increases pyruvate dehydrogenase kinase (PDK) activity, which phosphorylates and inactivates the pyruvate dehydrogenase complex (PDC), shrinking the lipoylated target pool in mitochondria and cutting the feed into the TCA cycle. Second, glycolytic signalling suppresses cuproptosis-promoting genes such as FDX1 and dihydrolipoamide S-acetyltransferase while inducing the negative regulator glutaminase (GLS), further lowering copper sensitivity. Third, diversion of glycolytic intermediates into the pentose-phosphate pathway (PPP) supplies abundant nicotinamide adenine dinucleotide phosphate (NADPH), whereas enhanced glutamine catabolism furnishes glutamate; together these fuels expand reduced glutathione (GSH) and metallothionein (MT) pools that chelate Cu¹⁺ and quench reactive oxygen species exactly where cuproptosis is executed. Consequently, glycolysis-dependent cancer cells are far less sensitive to copper-ionophore drugs such as elesclomol or disulfiram than respiration-dependent counterparts, and clinical datasets consistently link high PDK and low PDC-subunit expression with poor prognosis. These insights highlight rational combination strategies: re-activating the TCA cycle with PDK inhibitors, draining PPP- or GLS-driven NADPH/GSH supply, and concurrently delivering copper ionophores could reopen the cuproptotic trap in tumours. Validating such approaches in vivo, charting upstream regulators of FDX1 and mapping crosstalk between cuproptosis and other lethal programmes remain key steps toward exploiting this copper-centred vulnerability in cancer therapy.</p>\",\"PeriodicalId\":72398,\"journal\":{\"name\":\"Biomolecules & biomedicine\",\"volume\":\" \",\"pages\":\"251-261\"},\"PeriodicalIF\":0.0000,\"publicationDate\":\"2025-08-06\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12505534/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Biomolecules & biomedicine\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.17305/bb.2025.12751\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"0\",\"JCRName\":\"MEDICINE, RESEARCH & EXPERIMENTAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biomolecules & biomedicine","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.17305/bb.2025.12751","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"0","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
Tumor glucose reprogramming suppresses cuproptosis: A review.
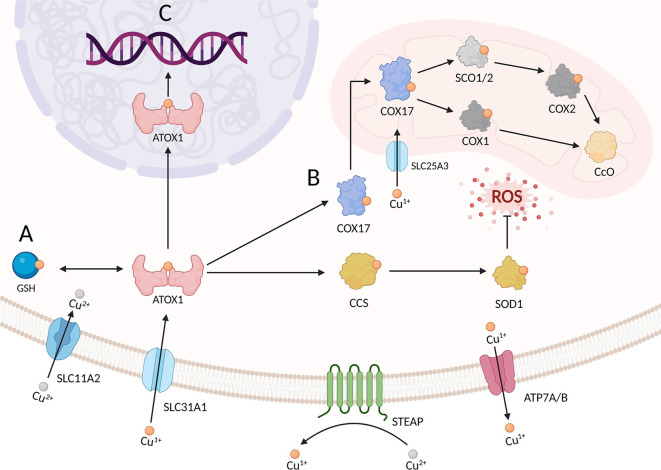
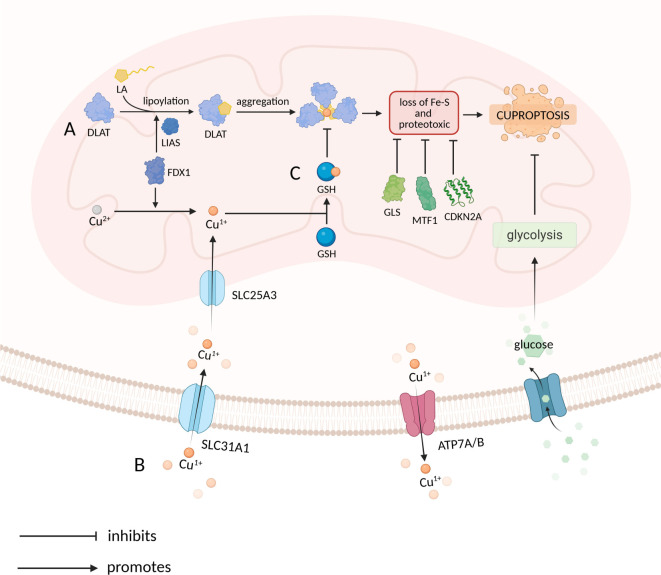
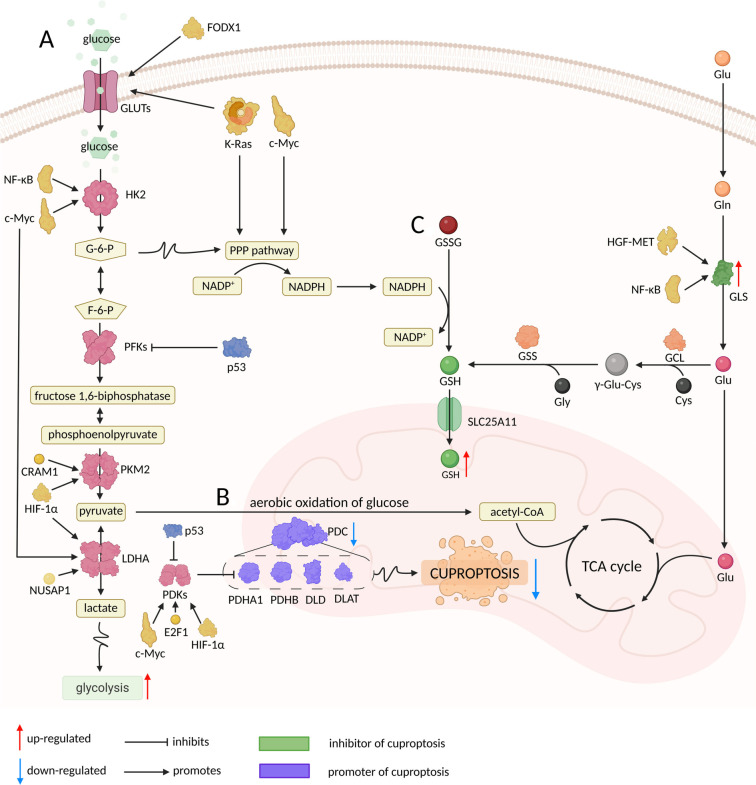
Cuproptosis is a copper-dependent form of regulated cell death that begins when ferredoxin 1 (FDX1) reduces Cu²⁺ to Cu¹⁺, allowing the ion to bind lipoylated enzymes of the tricarboxylic-acid (TCA) cycle, drive protein aggregation, dismantle iron-sulphur clusters and trigger fatal proteotoxic stress. Most tumours, despite accumulating copper, evade this fate through glucose-metabolic rewiring. First, oncogenic stabilisation of hypoxia-inducible factor-1 alpha (HIF-1α) and MYC increases pyruvate dehydrogenase kinase (PDK) activity, which phosphorylates and inactivates the pyruvate dehydrogenase complex (PDC), shrinking the lipoylated target pool in mitochondria and cutting the feed into the TCA cycle. Second, glycolytic signalling suppresses cuproptosis-promoting genes such as FDX1 and dihydrolipoamide S-acetyltransferase while inducing the negative regulator glutaminase (GLS), further lowering copper sensitivity. Third, diversion of glycolytic intermediates into the pentose-phosphate pathway (PPP) supplies abundant nicotinamide adenine dinucleotide phosphate (NADPH), whereas enhanced glutamine catabolism furnishes glutamate; together these fuels expand reduced glutathione (GSH) and metallothionein (MT) pools that chelate Cu¹⁺ and quench reactive oxygen species exactly where cuproptosis is executed. Consequently, glycolysis-dependent cancer cells are far less sensitive to copper-ionophore drugs such as elesclomol or disulfiram than respiration-dependent counterparts, and clinical datasets consistently link high PDK and low PDC-subunit expression with poor prognosis. These insights highlight rational combination strategies: re-activating the TCA cycle with PDK inhibitors, draining PPP- or GLS-driven NADPH/GSH supply, and concurrently delivering copper ionophores could reopen the cuproptotic trap in tumours. Validating such approaches in vivo, charting upstream regulators of FDX1 and mapping crosstalk between cuproptosis and other lethal programmes remain key steps toward exploiting this copper-centred vulnerability in cancer therapy.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


