N van Engelen, H M van Santen, F van Dijk, M M Kleisman, J H M Merks, A Y N Schouten-van Meeteren, E J Kamping, K Neveling, A Hoischen, M C J Jongmans, R P Kuiper
{"title":"利用全基因组测序和光学基因组作图对5例ROHHAD-NET患者进行遗传评价。","authors":"N van Engelen, H M van Santen, F van Dijk, M M Kleisman, J H M Merks, A Y N Schouten-van Meeteren, E J Kamping, K Neveling, A Hoischen, M C J Jongmans, R P Kuiper","doi":"10.1186/s13023-025-03938-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Rapid-onset obesity, hypothalamic dysfunction, hypoventilation, autonomic dysregulation (ROHHAD) and neuroendocrine tumor (NET) is a very rare condition with an unknown etiology. While various potential causes have been hypothesized, including genetic and paraneoplastic autoimmune mechanisms, no definitive cause has been identified to date. This study aimed to explore whether patients with ROHHAD-NET share an underlying heritable genetic etiology.</p><p><strong>Results: </strong>We identified five female patients clinically suspected of having ROHHAD(-NET); among them in two patients a NET was found: a ganglioneuroma and a low grade cerebellar ganglion cell tumor with BRAF mutation. To identify potential pathogenic germline genomic variants, whole genome sequencing (WGS) was performed on germline DNA from all five patients, including four patient-parent trios. Furthermore, optical genome mapping (OGM) was performed for two patients to detect germline structural variants (SVs). Rare single nucleotide variants (SNVs) and small insertions/deletions (InDels) were identified through WGS and rare SVs affecting (non)-coding or regulatory regions were analyzed using both WGS and OGM. We explored a de novo, inherited autosomal dominant and autosomal recessive inheritance scenario. However, no candidate variants in a recurrently affected gene locus or genomic region were identified in two or more patients.</p><p><strong>Conclusion: </strong>Our comprehensive genome-wide data analysis did not reveal evidence of a monogenetic cause for ROHHAD-NET. Whereas these findings do not exclude a genetic etiology for ROHHAD-NET, they strengthen the hypothesis of an autoimmune origin for symptoms of ROHHAD.</p>","PeriodicalId":19651,"journal":{"name":"Orphanet Journal of Rare Diseases","volume":"20 1","pages":"412"},"PeriodicalIF":3.5000,"publicationDate":"2025-08-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12333276/pdf/","citationCount":"0","resultStr":"{\"title\":\"Genetic evaluation of five patients with ROHHAD-NET using whole genome sequencing and optical genome mapping.\",\"authors\":\"N van Engelen, H M van Santen, F van Dijk, M M Kleisman, J H M Merks, A Y N Schouten-van Meeteren, E J Kamping, K Neveling, A Hoischen, M C J Jongmans, R P Kuiper\",\"doi\":\"10.1186/s13023-025-03938-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Rapid-onset obesity, hypothalamic dysfunction, hypoventilation, autonomic dysregulation (ROHHAD) and neuroendocrine tumor (NET) is a very rare condition with an unknown etiology. While various potential causes have been hypothesized, including genetic and paraneoplastic autoimmune mechanisms, no definitive cause has been identified to date. This study aimed to explore whether patients with ROHHAD-NET share an underlying heritable genetic etiology.</p><p><strong>Results: </strong>We identified five female patients clinically suspected of having ROHHAD(-NET); among them in two patients a NET was found: a ganglioneuroma and a low grade cerebellar ganglion cell tumor with BRAF mutation. To identify potential pathogenic germline genomic variants, whole genome sequencing (WGS) was performed on germline DNA from all five patients, including four patient-parent trios. Furthermore, optical genome mapping (OGM) was performed for two patients to detect germline structural variants (SVs). Rare single nucleotide variants (SNVs) and small insertions/deletions (InDels) were identified through WGS and rare SVs affecting (non)-coding or regulatory regions were analyzed using both WGS and OGM. We explored a de novo, inherited autosomal dominant and autosomal recessive inheritance scenario. However, no candidate variants in a recurrently affected gene locus or genomic region were identified in two or more patients.</p><p><strong>Conclusion: </strong>Our comprehensive genome-wide data analysis did not reveal evidence of a monogenetic cause for ROHHAD-NET. Whereas these findings do not exclude a genetic etiology for ROHHAD-NET, they strengthen the hypothesis of an autoimmune origin for symptoms of ROHHAD.</p>\",\"PeriodicalId\":19651,\"journal\":{\"name\":\"Orphanet Journal of Rare Diseases\",\"volume\":\"20 1\",\"pages\":\"412\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2025-08-07\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12333276/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Orphanet Journal of Rare Diseases\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13023-025-03938-3\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Orphanet Journal of Rare Diseases","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13023-025-03938-3","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Genetic evaluation of five patients with ROHHAD-NET using whole genome sequencing and optical genome mapping.
Background: Rapid-onset obesity, hypothalamic dysfunction, hypoventilation, autonomic dysregulation (ROHHAD) and neuroendocrine tumor (NET) is a very rare condition with an unknown etiology. While various potential causes have been hypothesized, including genetic and paraneoplastic autoimmune mechanisms, no definitive cause has been identified to date. This study aimed to explore whether patients with ROHHAD-NET share an underlying heritable genetic etiology.
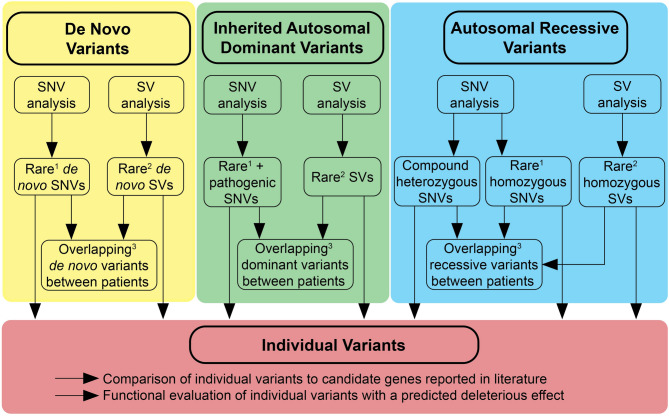
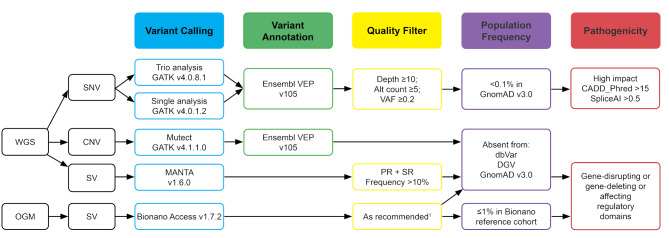
Results: We identified five female patients clinically suspected of having ROHHAD(-NET); among them in two patients a NET was found: a ganglioneuroma and a low grade cerebellar ganglion cell tumor with BRAF mutation. To identify potential pathogenic germline genomic variants, whole genome sequencing (WGS) was performed on germline DNA from all five patients, including four patient-parent trios. Furthermore, optical genome mapping (OGM) was performed for two patients to detect germline structural variants (SVs). Rare single nucleotide variants (SNVs) and small insertions/deletions (InDels) were identified through WGS and rare SVs affecting (non)-coding or regulatory regions were analyzed using both WGS and OGM. We explored a de novo, inherited autosomal dominant and autosomal recessive inheritance scenario. However, no candidate variants in a recurrently affected gene locus or genomic region were identified in two or more patients.
Conclusion: Our comprehensive genome-wide data analysis did not reveal evidence of a monogenetic cause for ROHHAD-NET. Whereas these findings do not exclude a genetic etiology for ROHHAD-NET, they strengthen the hypothesis of an autoimmune origin for symptoms of ROHHAD.
期刊介绍:
Orphanet Journal of Rare Diseases is an open access, peer-reviewed journal that encompasses all aspects of rare diseases and orphan drugs. The journal publishes high-quality reviews on specific rare diseases. In addition, the journal may consider articles on clinical trial outcome reports, either positive or negative, and articles on public health issues in the field of rare diseases and orphan drugs. The journal does not accept case reports.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


